Last edited: 08.24 .2019
Outline.
- Glycogen Metabolism
- Fatty Acid Metabolism
- Ketone Body Metabolism
- Amino Acid & Nitrogen Metabolism
Glycogen.
- Glycogen is stored in the cytosolic granules of the muscle and liver cells.
- Glycogen is important to the body particularly for the CNS because the CNS is an obligate user of glucose.
- IN THE LIVER, glycogen is broken down or stored in order to maintain the correct balance of blood sugar. There’s about 100g or about 10% glycogen in fresh weight well fed adult liver.
- IN THE LIVER, the enzyme that transforms glucose into glucose 6-phosphate is called glucokinase.
- IN THE MUSCLES, glycogen is used for energy especially for anaerobic high intensity bouts. There’s about 400g glycogen or 1-2% of fresh weight muscle.
- IN THE MUSCLES, the enzyme that transforms glucose into glucose 6-phosphate is hexokinase.
- Glycogen is a branched-chain polymer made up of glucose monomers linked up via alpha-1,4-glycosidic bonds in a straight chain, and alpha-1,6-glycosidic bonds at branches.
- There are more alpha-1,4-glycosidic bonds than the alpha-1,6 bonds.
- After about 8-14 glucose residues, a branch occurs.
- Nonreducing ends. The 1,4-glycosidic bonds dominate the structure and their “ends” have a free hydroxl group at carbon 4. These are called the nonreducing ends.
- Reducing end. The rightmost oriented glucose has a free anomeric carbon and is called the reducing end. This reducing end is usually attached to a protein (glycogenin).
- Reducing sugars: sugars that can freely open and close their ring-form.
- Is the anomeric carbon free? If something is bonded to the sugar at the anomeric carbon (carbon 1) this “blocks” the sugar from freely opening and closing its ring form. Thus the sugar is not reducible.
- When studying glycogen, a protein is often bound to the glycogen at the carbon 1 position. This improves stability as the sugar cannot freely “shift” between open- and closed-ring forms.
- Glycogenin: is an enzyme/protein that serves as the “head” or “anchor” where glycogen chains and branches can attach. Glycogenin is attached to the first glucose in a glycogen chain. Glycogenin initiates glycogen synthesis. Afterwards, glycogen synthase takes over.
- Futile cycling: aka substrate cycling; substrate gets converted into product via one pathway and then the product gets converted back to the substrate in another pathway.
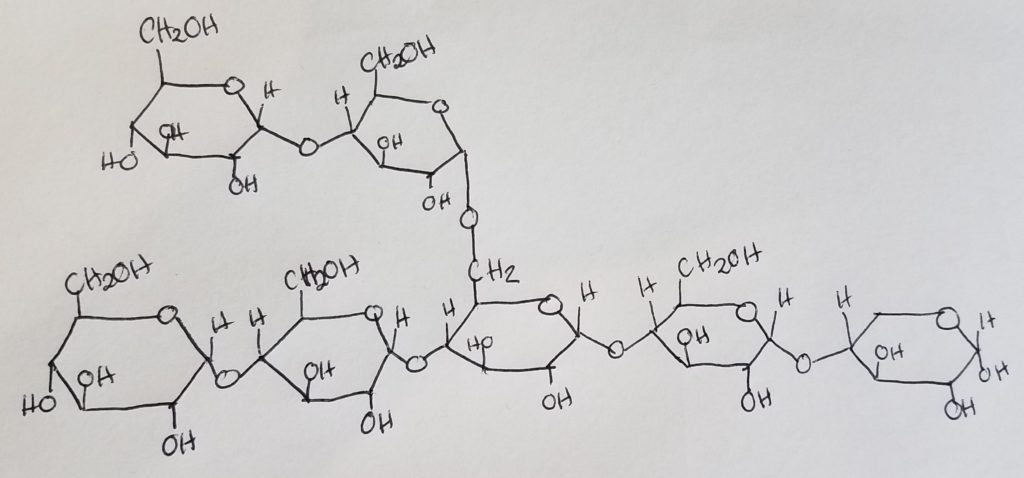
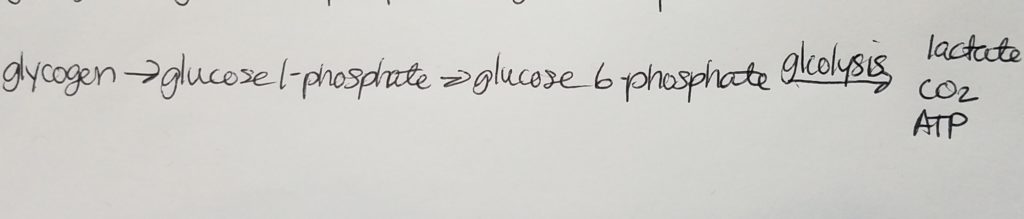

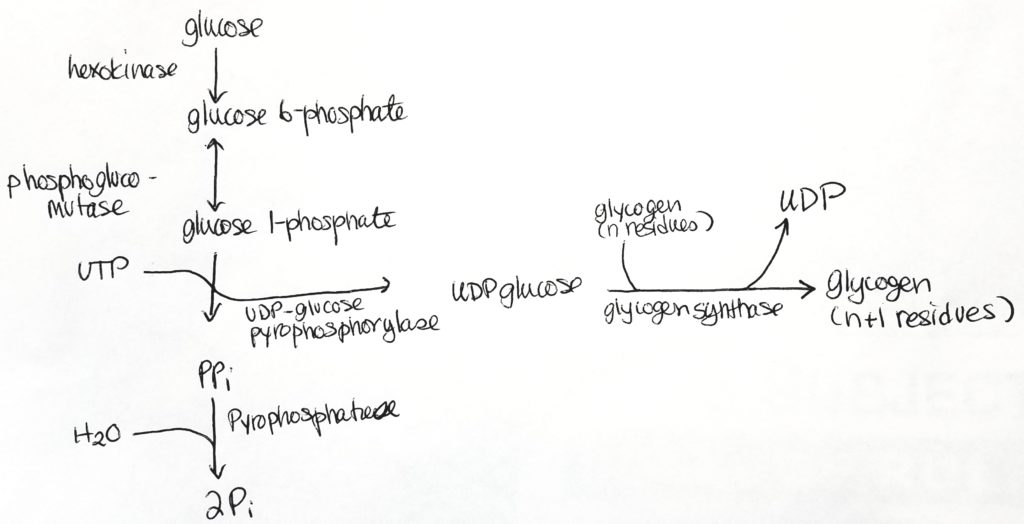
Glycogenesis (Glycogen synthesis).
- Glycogenesis occurs in the cytosol.
- Glycogen is made from alpha-D-glucose.
- ATP and UTP (uridine triphosphate) are used for energy in this process.
- 3 enzymatic steps convert G6P into glycogen.
- Glycogen synthase is the major regulatory step. It makes alpha-1,4 bonds.
- UTP/UDP are carriers.
- Mutase is a special form of isomerase. Mutases take a functional group at one position and moves it to another position.
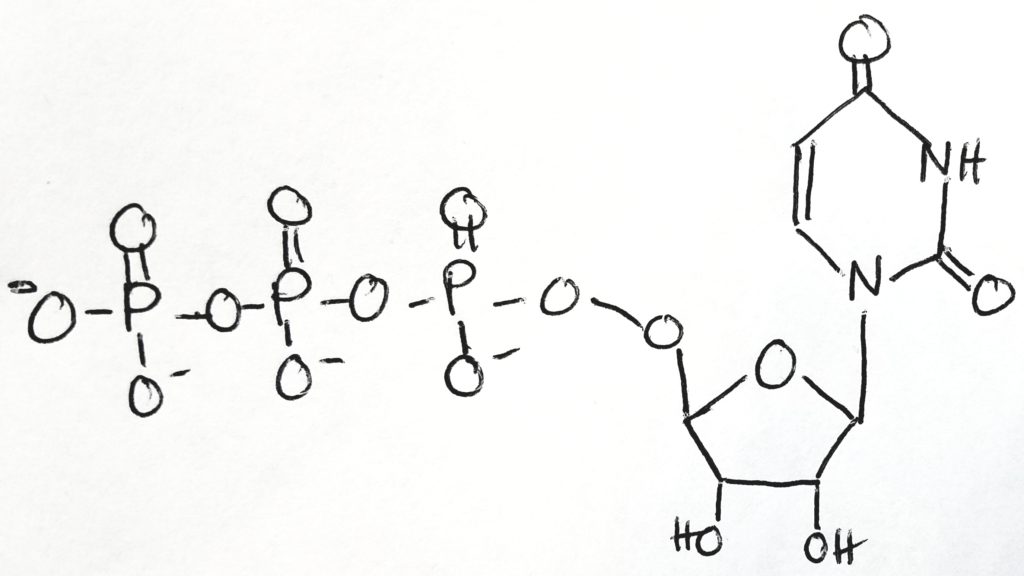
Uridine-5′-triphosphate ( C9H15N2O15P3 ).
Step 1.
- In the liver, glucose-6-phosphatase may cleave a glucose monomer of glucose 6-phosphate so that a glucose molecule may enter the bloodstream and be used by cells for energy.
- glucose 6-phosphate + H2O –glucose-6-phosphatase–> glucose + Pi
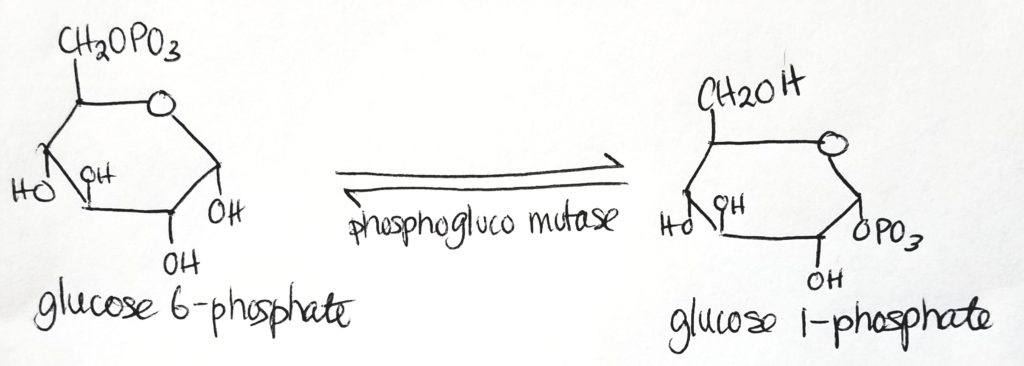
Step 2.
- UDP-glucose is the activated form.
- UDP-glucose is the form which is used directly to form glycogen.
- Glucose 1-phosphate has one phosphate group.
- Take UTP, free two phosphates from it as PPi.
- Then attach UMP (monophosphate) to glucose 1-phosphate to get UDP-glucose (which now has 2 phosphate groups, one from previously and one from the UTP).
- Cleavage of the phosphate bonds give enough energy to attach monomers to the glycogen chain.
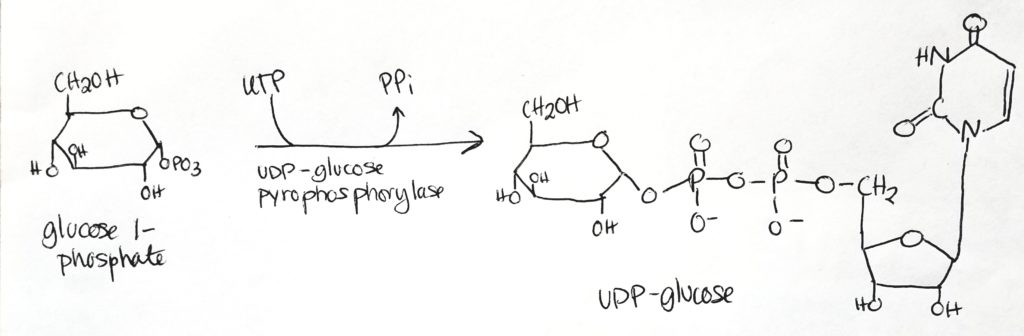
Step 3.
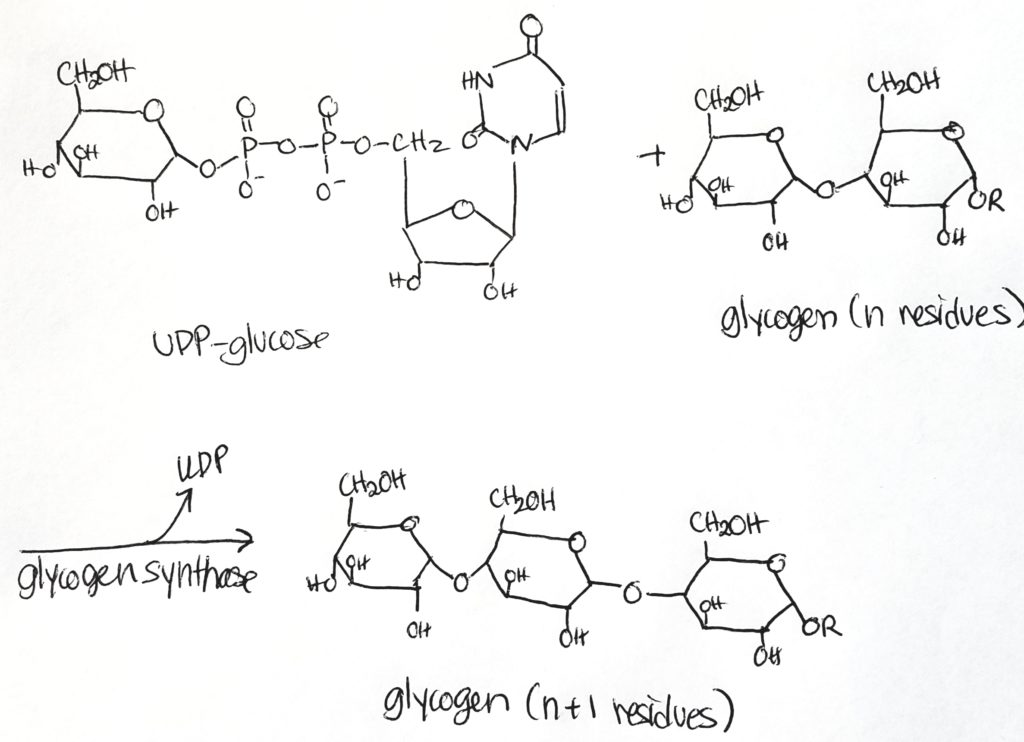
Glycogen Synthesis Enzymes.
- Glycogenin. Gets the process started and then glycogen synthase takes over. Glycogenin: is an enzyme/protein that serves as the “head” or “anchor” where glycogen chains and branches can attach. Glycogenin is attached to the first glucose in a glycogen chain. Glycogenin initiates glycogen synthesis. Afterwards, glycogen synthase takes over.
- Glycogen synthase. Adds a glucose monomer to the chain (elongation process). Creates alpha-1,4-linkages only. Adds one glucose unit to an existing chain of at least 8 molecules (elongation).
- 1,4-Alpha-Glucan Branching enzyme. Takes 6-8 residues at a time and forms a new branching point. Creates alpha-1,6-linkages only. Branching is important in that it creates a lot more terminals and potential sites for phosphorylation and synthesis (make it easier to multitask by increasing more sites/terminals). Also increases solubility by making more “pockets” for water molecules to get into.
- Putting a phosphate group onto something can alter it’s function and activate or deactivate it.
- Rarely do we say “turn on” or “turn off”, because it’s not that black and white.
- Glycogen phosphorylase. It is a dimer with quaternary structure. It has one copy “above” and one copy”below”. Specifically targets alpha-1,4-linkages. It cleaves off a glucose 1-phosphate. Once a glycogen chain gets down to 4 units of glucose monomer, glycogen phosphorylase cannot work. That’s where the debranching enzyme comes in.
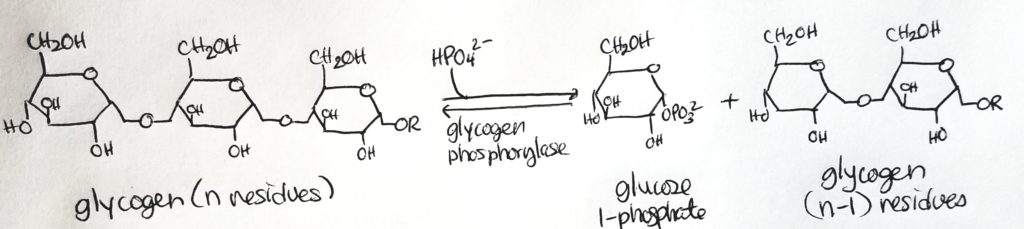
Glycogenolysis.
- Catabolic/breakdown of glycogen to glucose units so that the glucose can enter the bloodstream and be utilized or so that glucose can be utilized in the muscle.
- This process uses Pi to cleave a glucose from glycogen (it’s not a hydrolysis reaction). It takes energy to cleave that glucose out.
- This cleavage using Pi is called phosphorylation.
- Glycogen phosphorylase is the enzyme that catalyzes this phosphorylation.
Glycogen Debranching Enzyme.
- Another way to catabolize glycogen.
- Glycogen phosphorylase (previously studied) was able to cleave a terminal glucose unit off of glycogen. Note that this unit is in the form glucose 1-phosphate.
- Glycogen phosphorylase CANNOT act on any branches that are less than 5 glucose monomers long.
- Glycogen debranching enzyme is a hydrolase (use water to cleave) NOT phosphorylase (using phosphate to cleave).
- Glucose is the result of cleavage and NOT glucose 1-phosphate.
- This enzyme has both a 4:4 transferase activity and a glucosidase activity.
- Once a branch has been pruned such that there are only 4 glucose units on it, the debranching enzyme (the 4:4 transferase role) will prune the 3 terminal units of glucose from that branch and attach them to a longer branch/chain.
- Then the debranching enzyme (glucosidase role) will prune off that lone glucose unit, making it available in the cell/bloodstream. Note that it is glucose and NOT glucose 1-phosphate or glucose 6-phosphate. It’s a straightup glucose.
- Note that the glucosidase role of the debranching enzyme can only act to prune off a glucose unit that is 1 unit long. For example, if there is a branch of 2 glucose units, the debranching enzyme cannot act on it.
- Note that the glucosidase role is a hydrolase. It uses water to cleave the glucose unit and set it free.
Regulation of Glycogen Metabolism.
- Glycogen synthase and glycogen phosphorylase help regulate glycogen metabolism.
- Both are allosteric regulators and respond to hormonal control (via phosphorylation and dephosphorylation).
- Glycogen synthase is activated by glucose 6-phosphate (indicating glucose levels are high).
- Glycogen phosphorylase is inhibited by high levels of glucose 6-phosphate and activated with high levels of AMP (indicating we need more energy).
Insulin.
- Insulin is a hormone that responds to high blood glucose levels.
- When blood glucose levels are high, insulin inhibits glycogen phosphorylase (don’t free up any more glucose because we don’t need it).
- Insulin activates glycogen synthase.
- Insulin is associated with the fed state.
Glucagon and Epinephrine.
- These are active when there is low blood glucose.
- These hormones inhibit glycogen synthase (we don’t want to store energy, we need energy to use now).
- These hormones activate glycogen phosphorylase.
- Associated with fasting state and/or exercise states.
Fatty Acid Metabolism (Oxidation).
- What are fatty acids (FAs)? They are a carboxylic acid head with a long alipathic (non-aromatic) hydrocarbon tail.
- What are fatty acids good for? Storage of energy; building blocks for other structures like phospholipid and glycolipids; used in eicosanoids structure and secondary messengers.
- Triacylglycerol = triglyceride = glycerol + 3 FA chains.
- Triacylglycerol is the simplest form of lipid.
- FAs are very reduced and have lots of potential for oxidation (great source of potential energy).
- FA oxidation produces NADH and FADH2 which can then be used in ETC to make lots of ATP.
- Fats are efficient energy storage.
- Carbohydrates = 4 kcal/g
- Fats = 9 kcal/g
- Advantages of fats over polysaccharides: FA has more energy per carbon because FAs are more reduced; FAs carry less water because they are nonpolar.
- Short-term energy, “quick” delivery: glucose and glycogen.
- Long-term energy, slower delivery: fats.
- 1/3 of our energy needs are from triacylglycerols.
- The CNS and red blood cells cannot use FA’s because they don’t have the machinery to process and utilize FAs.
Fatty Acid Breakdown.
- Bile salts. Made in the liver, trickle through bile ducts, and stored in the gallbladder until they are released into the duodenum. The gall bladder helps to concentrate the bile. Act as emulsifiers and keep fat from making large clumps of itself. As emulsifiers, help to increase the surface area of fat “droplets” so that enzymes can have an easier time to reach and act on the lipids. Without bile salts, enzymes may not be as efficient acting on large clumps of fats. Emulsifiers also help “fat droplets” move through the body better. Large fat clumps could potentially cause fatal blockages. Bile salts help to “solubilize” lipids/triacylglycerol.
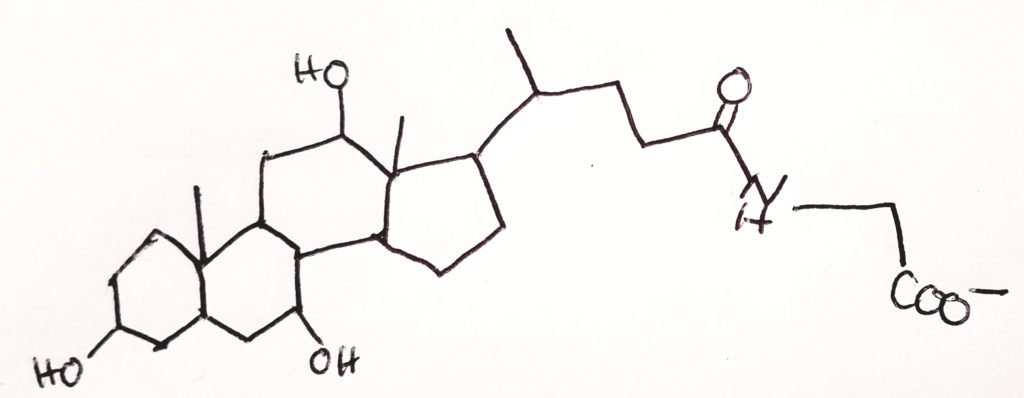
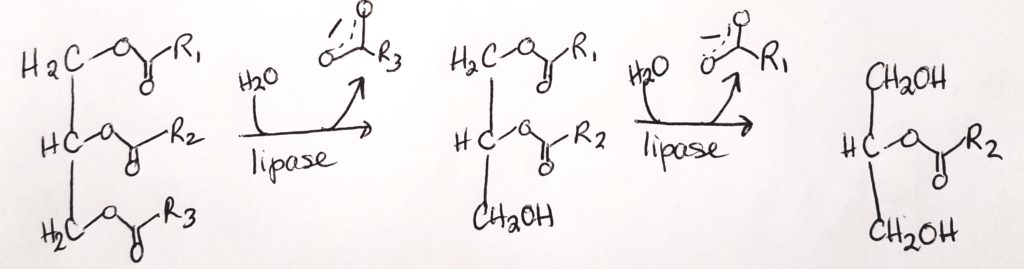
- Lipases. Lipases are made in the pancreas and they help to hydrolyze ester bonds (RCOOR) of triacylglycerols.
- FAs and two monoacylglycerols can then cross the plasma membrane of epithelial cells.
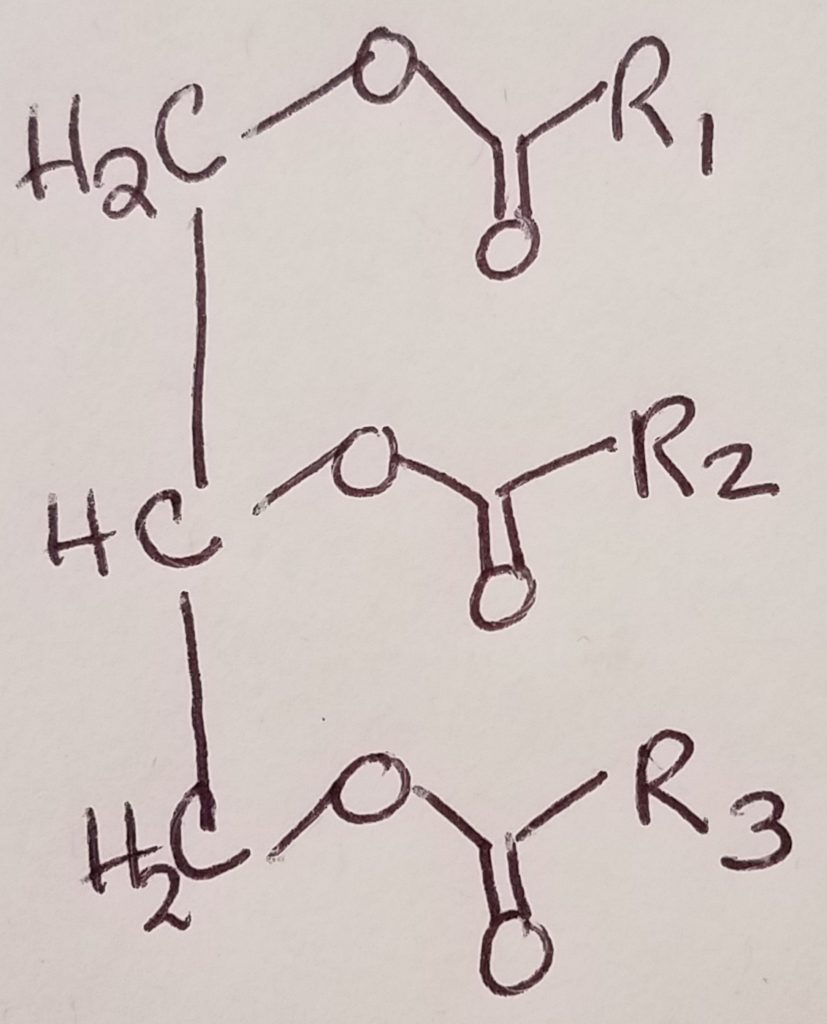
Triglyceride Digestion & Absorption.
- What kinds of lipids are in our diet? Triglycerides (triacylglycerols), fat-soluble vitamins, sterols (e.g. cholesterol), and “neutral fats”.
- What needs to happen in order for lipids to serve our needs? Because lipids are hydrophobic and generally insoluble in water, lipids physically need to be broken down into teeny tiny particles held in a suspension so that they can travel in the body which is largely an aqueous environment. Then enzymes need to act on the lipid particles in order to convert them to a form that the body can use/absorb.
- Digestion starts at the mouth.
- Mouth: lingual lipase initiates the breakdown of lipids/fats.
- Gall bladder – Liver: bile salts (amphipathic meaning they have both hydrophilic and hydrophobic qualities) are made in the liver and secreted/concentrated in the gall bladder. These bile salts are then released in the duodenum.
- Pancreas: secretes HCO3-, lipase colipase (a protein coenzyme that aids the pancreatic lipase). Pancreatic lipase is largely responsible for the bulk of breaking down lipids into tiny particles. Pancreatic lipase is also water soluble.
- Down in the stomach, the gastric lipase helps to break triacylglycerols into diacylglycerols…then break those down into fatty acid chains. The stomach churning and movement down into and through the intestines help keep the lipids emulsified (along with the bile salts, pancreatic lipase and colipase).
- The bile salts help to surround the fats to form micelles–fatty acid core with a more water-soluble “shell” or exterior. Bile salts are like detergents.
- Intestinal cells (enterocytes), are able to absorb the FAs and monoacylglycerols via FA protein transporter (to cross the cell membrane). The monoacylglycerols and fatty acids reassemble to form triacylglycerols. They are then attached to a protein carrier (lipoprotein).
- Inside the cell, the FAs and monoacylglycerols goto the endoplasmic reticulum so that they can be used to build triglycerides (triacylglycerols).
- From the ER to the Golgi Complex (of the intestinal cell, enterocyte) in assembly-line fashion, the triglycerides are packaged with cholesterol and lipoproteins into what’s called a chylomicrons.
- This combination of lipoproteins (1-2%), cholesterol (1-3%), phospholipid (6-12%), and core of triacylglycerols (85-92%) result in what is called a chylomicron (like a “super-sized” micelle). Chylomicrons exit the enterocyte via exocytosis and can pass into the lymphatic system and out into the bloodstream. Once in the bloodstream, the chylomicrons can disassemble to be used.
- Hormones can signal for lipolysis in adipose tissues: epinephrine, glucagon, cortisol promote lipolysis.
- Insulin inhibits lipolysis.
- FAs are bound to serum albumin for transport around the body.
- The lipases break down chylomicrons into glycerol and FAs.
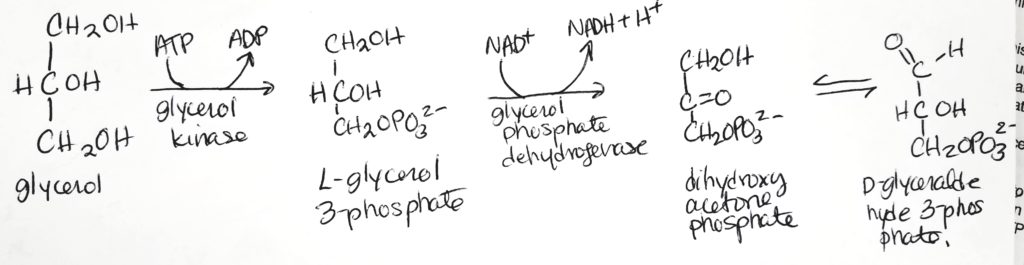
- Glycerol can get recycled to D-glyceraldehyde 3-phosphate which is an important intermediate for both glycolysis and gluconeogenesis.
Fatty Acid Activation.
- To oxidize FAs, first attach them to CoA and spend 2 ATP.
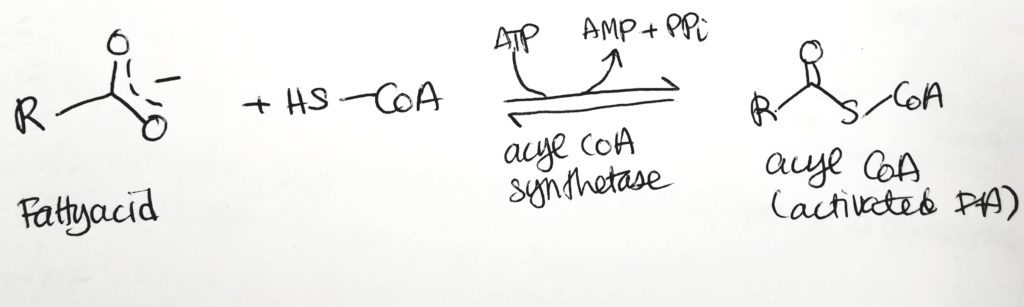
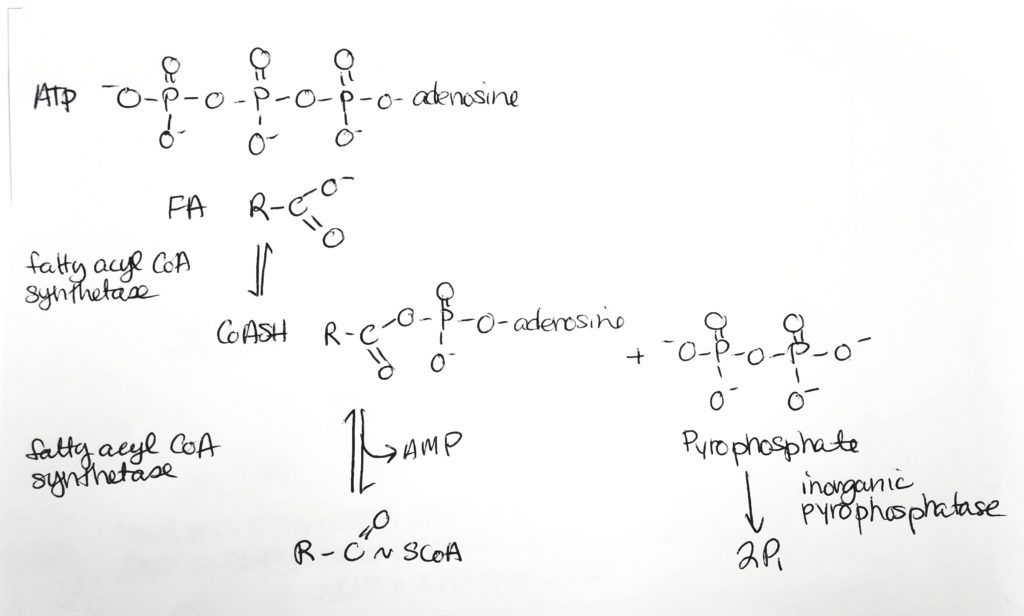
Getting the Fatty Acid into the Matrix.
- The fatty acyl CoA is shuttled via carnitine from the cytosol through the outer mitochondrial membrane (leaky) to the inner mitochondrial membrane.
- Carnitine palmitoyltransferase I (CPTI) enzyme is located on the outer mitochondrial membrane. It packages the fatty acyl CoA with carnitine.
- The fatty acyl CoA-carnitine is transported via carnitine translocase (enzyme) into the matrix.
- Carnitine palmitoyltransferase II (CPTII) located on the inner mitochondrial membrane then strips off the carnitine. The carnitine goes back out to the cytosol to be recycled. Then the fatty acyl CoA now present in the matrix can undergo beta-oxidation.
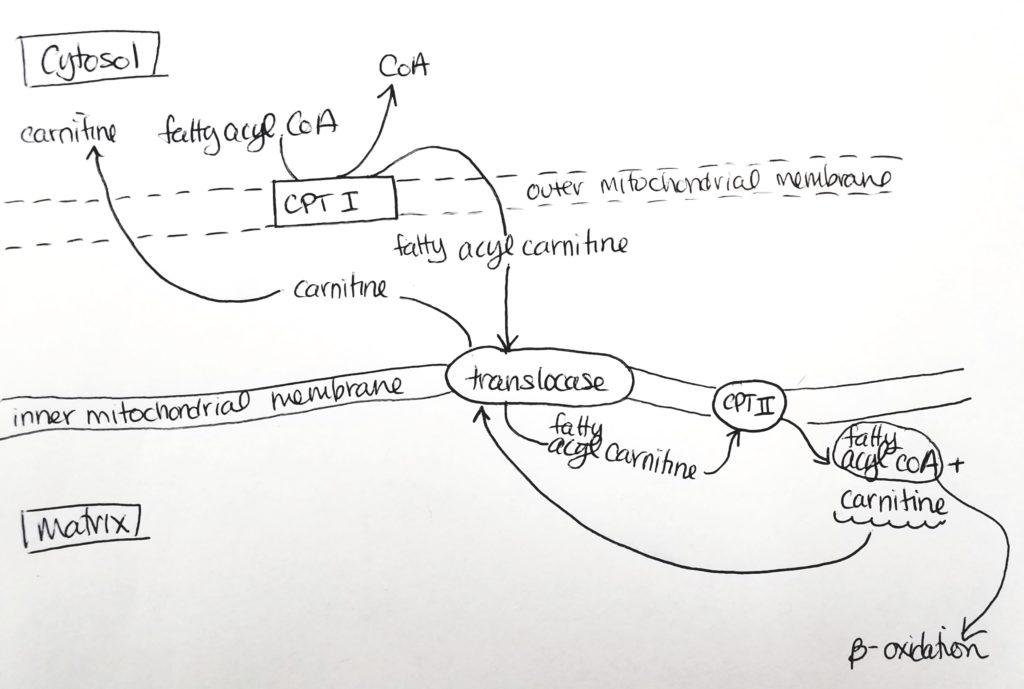
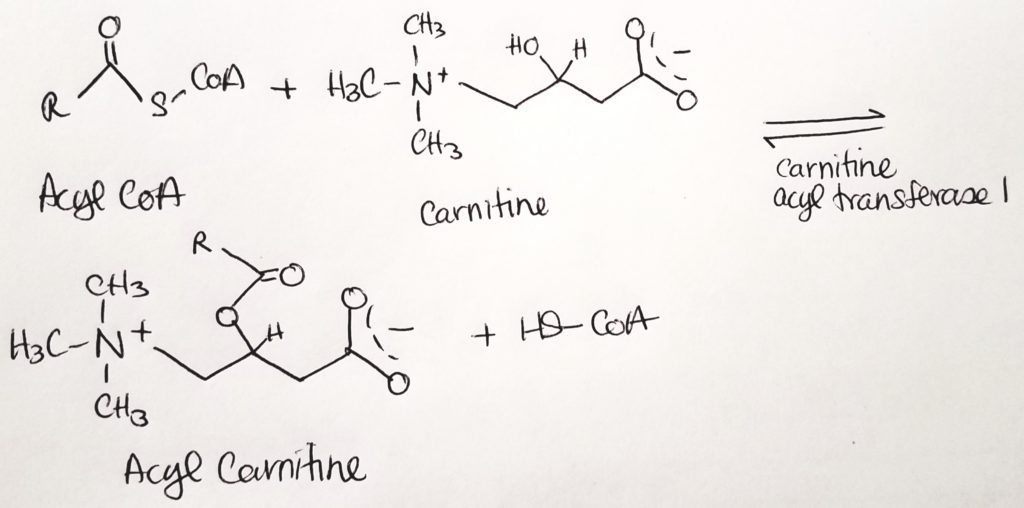
Beta-Oxidation.
- Beta-oxidation is so named because the activity is at the beta-carbon site.
- This is the catabolic pathway of fatty acids.
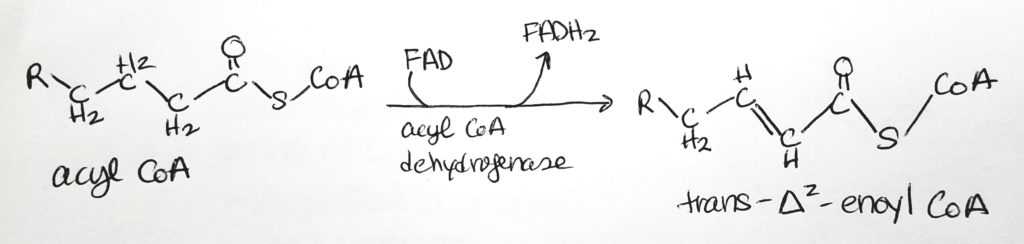
- Step 1. Oxidation. Makes 1 FADH2.
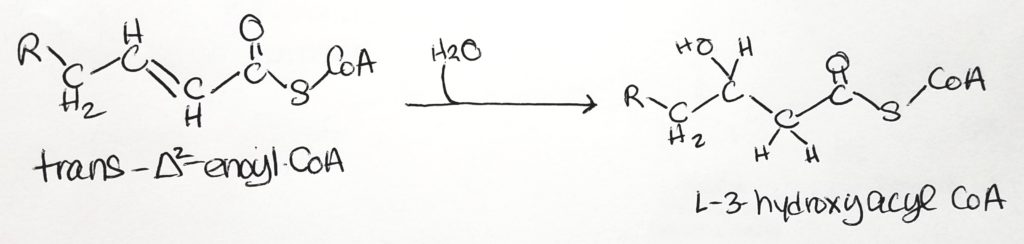
- Step 2. Hydration.
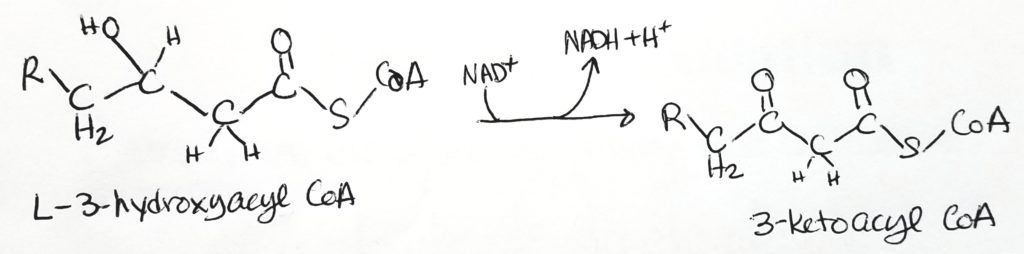
- Step 3. Oxidation. Makes NADH + H+
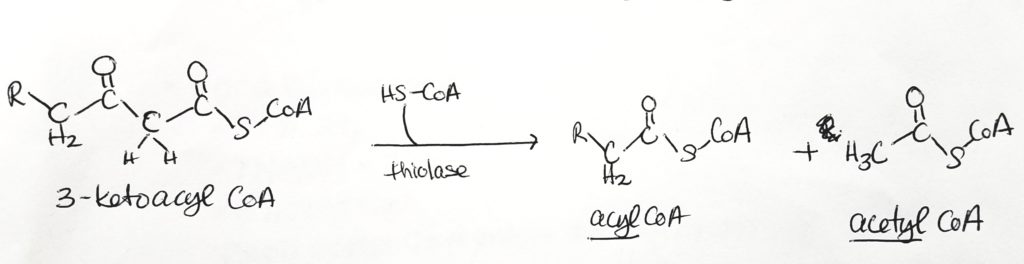
- Step 4. Thiolysis tomake one acyl CoA and one acetyl CoA
Calculating Energy Exchange of Fatty Acid Oxidation. What do you really get?
- Given n number of carbons.
- You have n/2 pairs of carbons.
- You cut the n long chain with (n/2)-1 many “snips”.
- You have (n/2)-1 many FADH2.
- You have (n/2)-1 many NADH and (n/2)-1 many H+
- You have n/2 many acetyl CoA.
- Each acetyl CoA will go through the TCA cycle to produce: 3 NADH, 1 FADH2, 1 GTP.
- You will go through n/2 rounds of the TCA cycle to produce: n/2 * 3 NADH; n/2 * 1 FADH2; n/2 * 1 GTP.
- FADH2 makes 1.5 ATP
- NADH makes 2.5 ATP
- 1 GTP makes 1 ATP
- As a shortcut you can think of each acetyl CoA that goes through the TCA makes 10 ATP.
What do you do with an odd-numbered FA chain?
Don’t panic. Odd numbered FA’s make propionyl-CoA. The last fragment of 3 carbons is called propionyl-CoA. HCO3- (bicarb) and ATP are added (coenzyme biotin). Coenzyme B12 is also active to make 4-carbon succinyl-CoA.
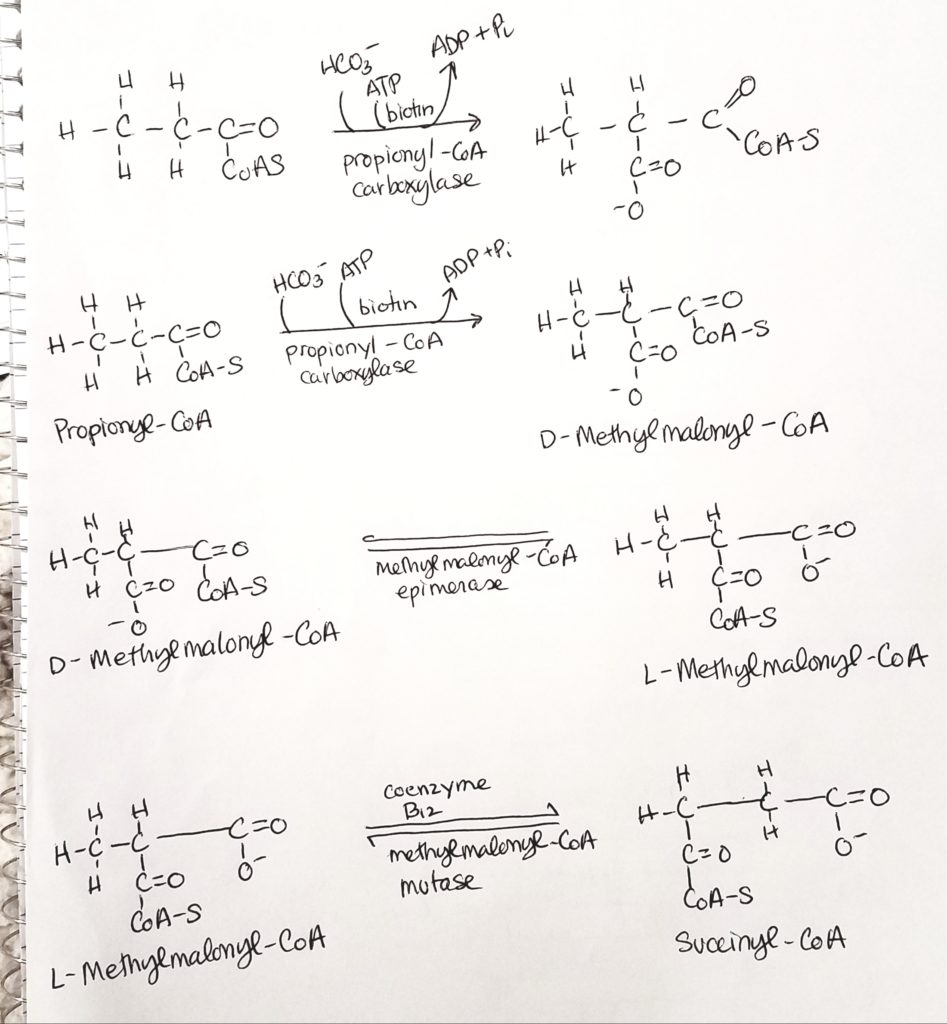
Let’s work an odd-numbered problem.
- Say you have a 17 carbon FA.
- (17/2) – 1.5 = 7 acetyl CoA.
- You can do the math for the 7 acetyl CoAs.
- REMEMBER to subtract the “cost” of 2 ATPs.
- But what’s left over?
- Propionyl undergoes carboxylations with biotin and B12 to produce succinyl-CoA.
- Add 5 ATP for the propionyl.
Fatty Acid Oxidation: Animals vs. Plants.
- Animals can’t use FAs to make glucose. Acetyl CoA can’t be converted to oxaloacetate.
- Plants have enzymes that let them convert acetyl CoA to oxaloacetate.
Ketone Body Metabolism.
- When lipid and carbohydrate metabolism are fairly balanced, most of the acetyl CoA from the FA beta-oxidation goes through the TCA cycle.
- Remember that the first step in the TCA cycle is oxaloacetate + water + acetyl CoA –citrate synthase–> citrate.
- BUT what if oxaloacetate supply is low? What do we do with the extra acetyl CoA?
- Oxaloacetate supply may be low if carbohydrates are scarce (starvation) or improperly used (misappropriated) such as diabetes.
- Oxaloacetate can be made from pyruvate via pyruvate carboxylase.
- Acetyl CoA drives ketone body production. When acetyl CoA levels persistantly rise, this triggers ketogenesis.
What are ketone bodies?
- Ketone bodies are 3 molecules produced in the mitochondrial matrix of hepatocytes (liver cells).
- These 3 molecules are: acetone, acetoacetate, and beta-hydroxybutyrate.
- The “making of” these 3 molecules is called ketogenesis. This occurs naturally in small amounts.
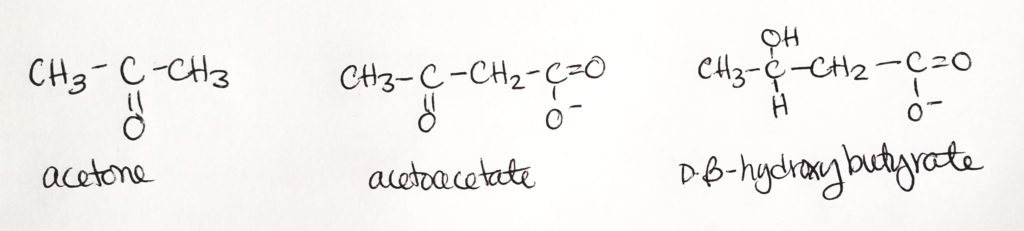
- When the body system is out of whack and acetone, acetoacetate, and/or D-beta-hydroxybutyrate are accumulated in eXcess, we call this state ketosis.
- When lots more ketone bodies accumulate in eXcess and the body’s pH is drastically lowered to acidic levels, we call this ketoacidosis.
Synthesis of Ketone Bodies.
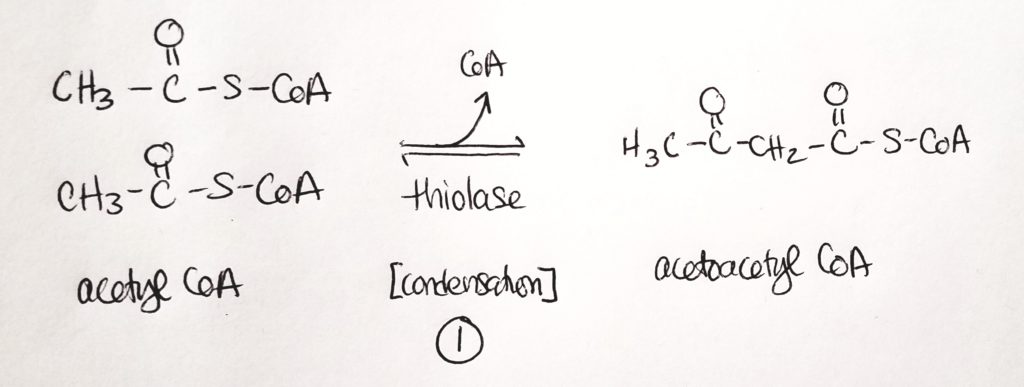
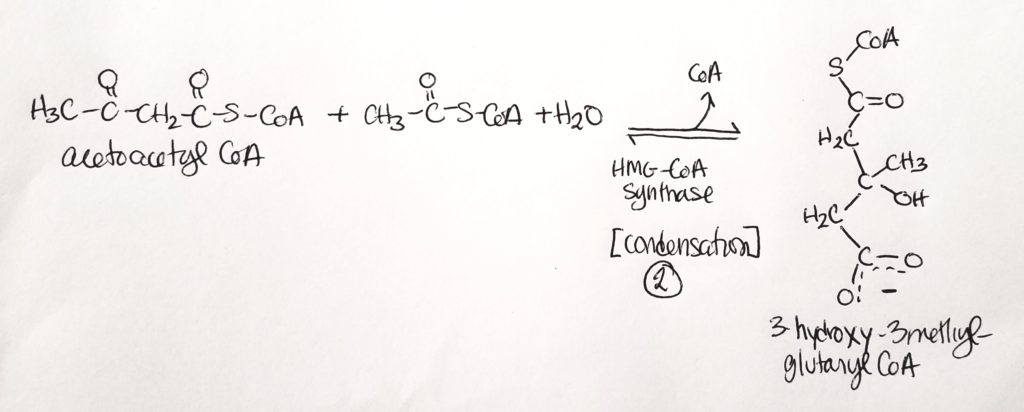
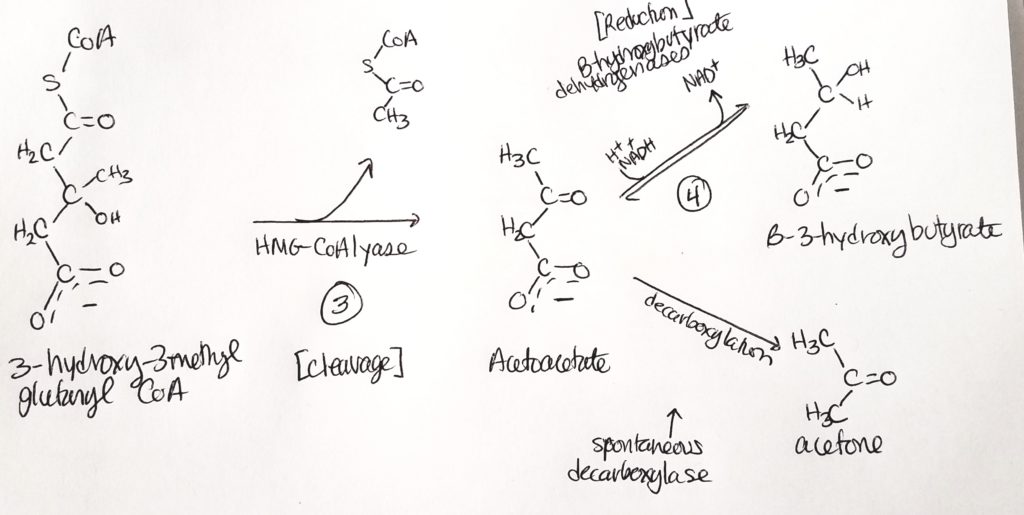
- B-3-hydroxybutyrate is the dominant product and is useful for a source of CoA.
- Step 1. Two acetyl CoA get joined into 1 acetoacetyl CoA. This is a condensation reaction. CoA is a leaving group (and one CoA stays).
- Step 2. Acetoacetyl CoA is joined to another acetyl CoA in a condensation reaction with water. One CoA is a leaving group.
- Step 3. One acetyl CoA is cleaved, removing the thiol. We’re left with a four-carbon molecule that B-hydroxybutarate dehydrogenase acts on.
- Step 4. End product is B-3-hydroxybutyrate.
- All this is going on in the mitochondrial matrix of hepatocytes (liver cells).
- So acetoacetate is spontaneously converted sometimes to acetone. Acetoacetate can also spend 1 NADH to form B-3-hydroxybutyrate.
- We can breathe acetone out (fruity breath).
- Acetoacetate and B-3-hydroxybutyrate can be peed out.
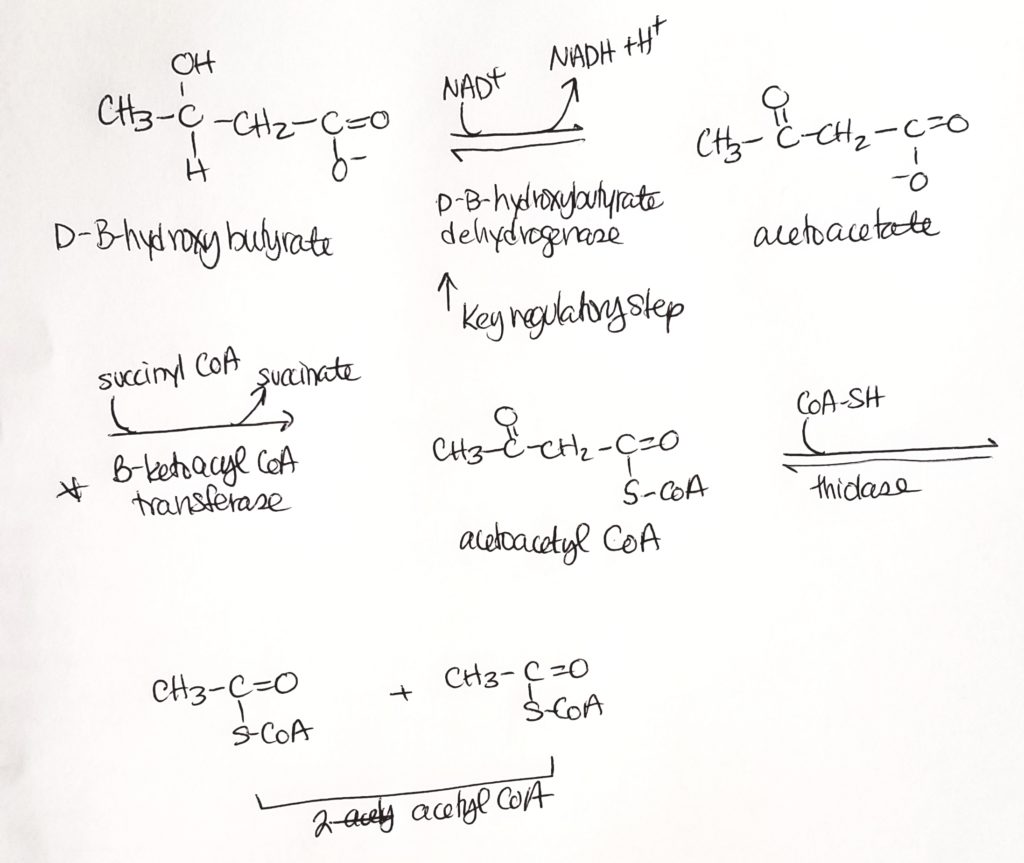
Ketone Body Oxidation (we made them, now we break them).
Amino Acid & Nitrogen Metabolism.
- Proteins are broken down into amino acids which gets absorbed in the small intestine via intestinal epithelial cells.
- Amino acids make proteins and other nitrogen-containing compounds.
- The nitrogens that aren’t used for building things get converted to urea and other nitrogenous waste products which get excreted and leave the body.
- Amino acids can make glucose.
- AA’s can be substrated for FA synthesis.
- Amino acids can also be used as a fuel source like the BCAA (branched chain amino acids) that many people use as supplements in muscle-building activities.
- Other nitrogen-containing compounds are: ATP, nucleotides, hormones, porphyrin rings.
- Key idea: how can we interconvert things, how many ways are there to interconvert things?
How to move Nitrogen around: Transamination.
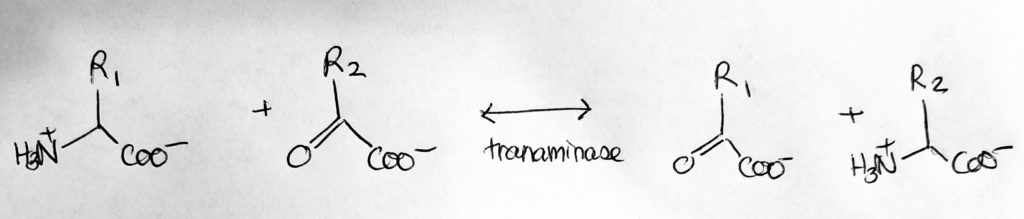
- Transamination. N-exchange.
- Catalyst: Transaminases/amino-transferases.
- Req: coenzyme PLP aka P5P (vitamin B6).
- Take the amine from one molecule and switch it with another group from another molecule. Turns amino acid to alpha-keto-acids and vice versa.
- All transamination reactions are reversible.
- Reactant-aminoacid1 becomes Product-alphaketoglutarate2; Reactant-alphaketoglutarate1 becomes Product-aminoacid2.
- Alanine <–> Pyruvate
- Aspartate <–> Oxaloacetate
- Alpha-Keto-Glutarate <–> Glutamate
- Asp + alphaketoglutarate <–asp aminotransferase, AST/GOT, PLP–> OA + Glu
- Ala + alphaketoglutarate <–Ala aminotransferase, ALT/GPT, PLP–> Pyr + Glu
- Leu + alphaketoglutarate <–Leu aminotransferase, LET/GOT, PLP–> OIC(oxoisocaproate) + Glu
How to move Nitrogen around: Amination.
- N-addition: +NH3
- Condensation reaction.
- Add nitrogen. For example, add +NH3.
- Aspartate + NH3 –> Asparagine
- Alpha-keto-glutarate + NH3 –> Glutamate
- Glutamate + NH3 –> Glutamine

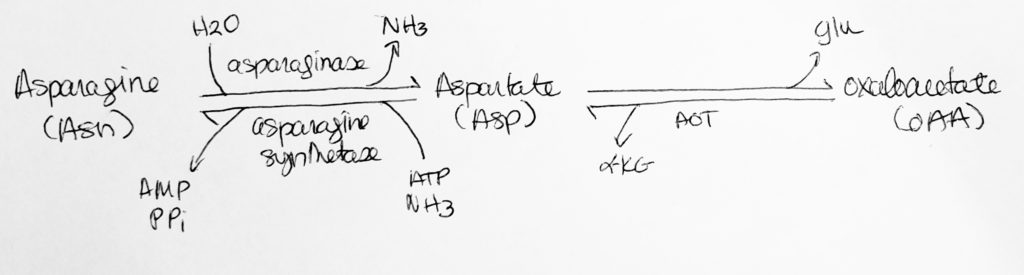
How to move Nitrogen around: Deamination.
- N-removal.
- Remove the nitrogen.
Fate of AA Carbons and Nitrogen.
- Carbons are used for energy.
- Nitrogen goes to the Urea cycle.
- One N in urea comes from NH4+ and the other N comes from Aspartate
Glucogenic & Ketogenic AA Degradation.
- Threonine > Glycine > [Alanine, Serine, or Cysteine] > Pyruvate.
- Tryptophan > [Alanine, Serine, or Cysteine] > Pyruvate.
- Pyruvate > Alanine.
- [Aspartate or Asparagine] > Oxaloacetate.
- [Aspartate, Tyrosine, or Phenylanine] > Fumarate.
- [Valine Threonine, Isoleucine, or Methionine] > Propionyl CoA.
- [Arginine, Histidine, Glutamine, or Proline] > Glutamate > Alpha-Keto-Glutarate.
- Leucine > HMG CoA
- [Phenylanine or Tyrosine] > Acetoacetate (ketone bodies)
- [Threonine, Lysine, Isoleucine, or Tryptophan] > Acetyl CoA
Urea (ornithine) Cycle.
- Happens only in the liver–part in liver cell’s mitochondria and part in liver cell’s cyctoplasm.
- One nitrogen comes from NH4+
- The OTHER nitrogen comes from Aspartate
- UREA is excreted (2x N).
Resources.
- Glycogen, http://watcut.uwaterloo.ca/webnotes/Metabolism/Glycogen.html
- Glycogen, http://www2.csudh.edu/nsturm/CHE452/04_Glycogen.htm
- Glycogen, https://academic.oup.com/nutritionreviews/article/76/4/243/4851715
- Glycogen, http://www.wou.edu/~guralnl/450Glycogen%20metabolism.pdf
- Glycogen, http://www.uh.edu/dtu/01-Glycogen-07.htm
- Glycogen, https://youtu.be/d-HgDkWK3P0
- Glycogen, https://youtu.be/P_5ubq6MikQ
- Glycogen I (OSU), https://youtu.be/Cuuy97TcBs0
- Glycogen II (OSU), https://youtu.be/GrxW9GXf6QU
- Glycogen, https://themedicalbiochemistrypage.org/glycogen.php#catabolism
- Glycogen, https://themedicalbiochemistrypage.org/glycogen.php#synthesis
- Glycogen, https://themedicalbiochemistrypage.org/glycogen.php#catabreg
- Glycogen, https://themedicalbiochemistrypage.org/glycogen.php#synthreg
- Glycogen, https://themedicalbiochemistrypage.org/glycogen.php#clinical
- Glycogen, https://themedicalbiochemistrypage.org/glycogen.php#storage
- Glycogen Debranching Enzyme, https://youtu.be/USCDwSmXpPU
- Fatty Acid, https://www.khanacademy.org/test-prep/mcat/biomolecules/fat-and-protein-metabolism/v/overview-of-fatty-acid-oxidation
- Fatty Acid, http://oregonstate.edu/instruct/bb451/451material/Keynotes/30FattyAcidMetabolism.pdf
- Fatty Acid, https://opentextbc.ca/anatomyandphysiology/chapter/24-4-lipid-metabolism/
- Fatty Acid, https://youtu.be/nBFSz63T1c0
- Fatty Acid, https://youtu.be/ooOx3G0JFi8
- Fatty Acid, https://youtu.be/__jS-pjzb5k
- Fatty Acid, https://youtu.be/ZufZvbhPpws
- Ketone, https://www.khanacademy.org/test-prep/mcat/biomolecules/fat-and-protein-metabolism/v/how-does-the-body-adapt-to-starvation
- Ketone, https://youtu.be/BlR9DkUAUkI
- Ketone, https://youtu.be/AK9Q2ClI8y4
- Ketone, https://youtu.be/IC9NmcaKX-w
- Ketone, https://www.rose-hulman.edu/~brandt/Chem330/Ketone_bodies.pdf
- Ketone, https://physoc.onlinelibrary.wiley.com/doi/full/10.1113/JP273185
- Ketone, https://www.dpag.ox.ac.uk/files/news/stubbs-b-et-al-front-physiol-oct-2017.pdf
- Ketone, https://www.researchgate.net/publication/313464452_Multi-dimensional_Roles_of_Ketone_Bodies_in_Fuel_Metabolism_Signaling_and_Therapeutics
- Amino acid, https://www.khanacademy.org/test-prep/mcat/biomolecules/fat-and-protein-metabolism/v/overview-of-amino-acid-metabolism
- Amino acid, http://watcut.uwaterloo.ca/webnotes/Metabolism/AminoAcids.html
- Amino acid, https://youtu.be/0M-B2dOfcUo
- Amino acid, https://youtu.be/EyHMLGKV4Wk
- Amino acid, https://www.youtube.com/channel/UCbYmF43dpGHz8gi2ugiXr0Q/search?query=amino+acid
- Nitrogen, https://media.oregonstate.edu/media/t/0_fbnugu4r
- Nitrogen, https://youtu.be/XmLzMCQ37iI
- Nitrogen, https://youtu.be/iNcglLxJ7mM
- Nitrogen, https://youtu.be/Y5xwfKdNS5s
- Nitrogen, https://youtu.be/XZEJ5PNvjkk
- Bile salts, http://www.vivo.colostate.edu/hbooks/pathphys/digestion/liver/bile.html
- Lipid digestion, https://med.libretexts.org/Bookshelves/Nutrition/Book%3A_An_Introduction_to_Nutrition_(Zimmerman)/05%3A_Lipids/5.4%3A_Digestion_and_Absorption_of_Lipids
- Carnitine Shuttle, https://med.libretexts.org/Bookshelves/Nutrition/Book%3A_Intermediate_Nutrition_(Lindshield)/6%3A_Macronutrient_and_Alcohol_Metabolism/6.3%3A_Lipid_Metabolism/6.32%3A_Fatty_Acid_Oxidation_(Beta-oxidation)
- Carnitine Shuttle, https://youtu.be/__jS-pjzb5k
- Carnitine Shuttle, https://youtu.be/ZufZvbhPpws
- The Odd Number Carbon, https://biochemistryquestions.wordpress.com/2008/12/14/beta-oxidation-of-fatty-acids-with-an-odd-number-of-carbons-i/
- Calculate the Even Number Carbon, https://youtu.be/oMnCckEvtBY
- Oxidation FA odd-number, https://youtu.be/_MiJLxjh-OE
References.
Bean, J. (2019). Amino acid nitrogen metabolism.
Bean, J. (2019). Fatty acid metaboslim.
Bean, J. (2019). Glycogen metaboslim.
Bean, J. (2019). Ketone body metabolism.
Ferrier, D. (2017). Biochemistry (7th ed.). Philidelphia, PA: Lippincott Illustrated Reviews.
Lieberman, M., & Peet, A. (2017). Marks’ basic medical biochemistry: A clinical approach(5th ed.). Philadelphia, PA: LWW.
Ferrier, D. (2017). Biochemistry (7th ed.). Philidelphia, PA: Lippincott Illustrated Reviews.
Lieberman, M., & Peet, A. (2017). Marks’ basic medical biochemistry: A clinical approach(5th ed.). Philadelphia, PA: LWW.
