Using Bio-Rad’s pGLO Bacterial Transformation Lab Kit (see Bio Rad’s link), our lab class made E. Coli glow in the dark.
See Bio-Rad’s Extensive Instructor and Student Documentation and Study Questions at this link [PDF]
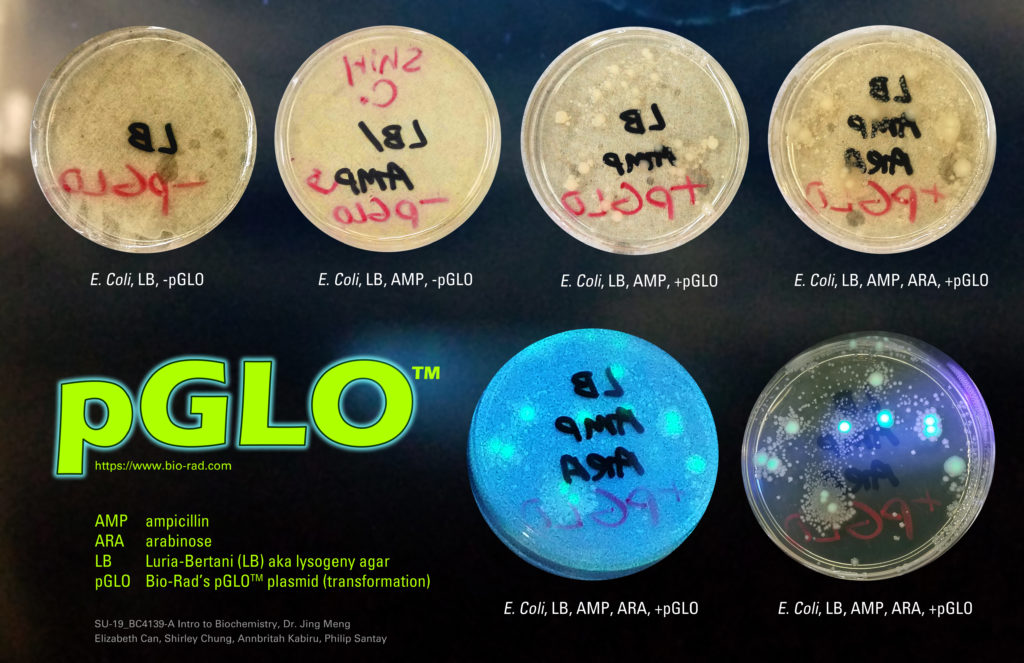
Using Bio-Rad’s pGLO Bacterial Transformation Lab Kit (see Bio Rad’s link), our lab class made E. Coli glow in the dark.
See Bio-Rad’s Extensive Instructor and Student Documentation and Study Questions at this link [PDF]
Last edited: 08.24 .2019
Outline.
Fatty Acid Synthesis.
| Characteristics | Synthesis | Degradation |
|---|---|---|
| Location | Cytosol | Mitochondrial Matrix |
| Activated Intermediates | Bound to ACP | Thioester of CoA |
| Intermediates | -SH of acyl carrier proteins | -SH of CoA |
| Activated Enzymes | FAS (multienzyme complex) | 4 distinct enzymes |
| Substrte | Acetyl CoA | Fatty Acids |
| Direction | Starts at methyl end | Starts at carboxyl end |
| Cofactors | NADPH/NADP+ | FADH2/NADH, FAD/NAD+ |
| Major Sites | Liver | Muscle & Liver |
| Hormonal Regulation | High Insulin/Glucagon Ratio | Low Insulin/Glucagon Ratio |
| Activator | Citrate | Free FA |
| Inhibitor | FA CoA | Malonyl CoA |
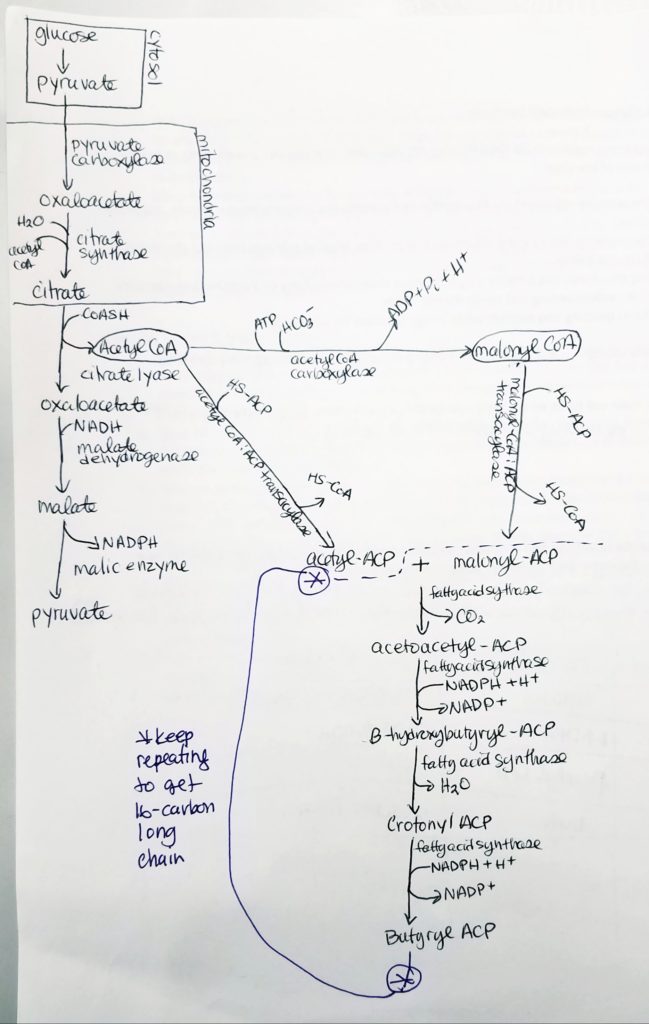
Acetyl CoA to Malonyl CoA.

Acyl Carrier Protein (ACP).
Step 0: Activation/Loading.


Fatty Acid Synthesis Step 1. Condensation.
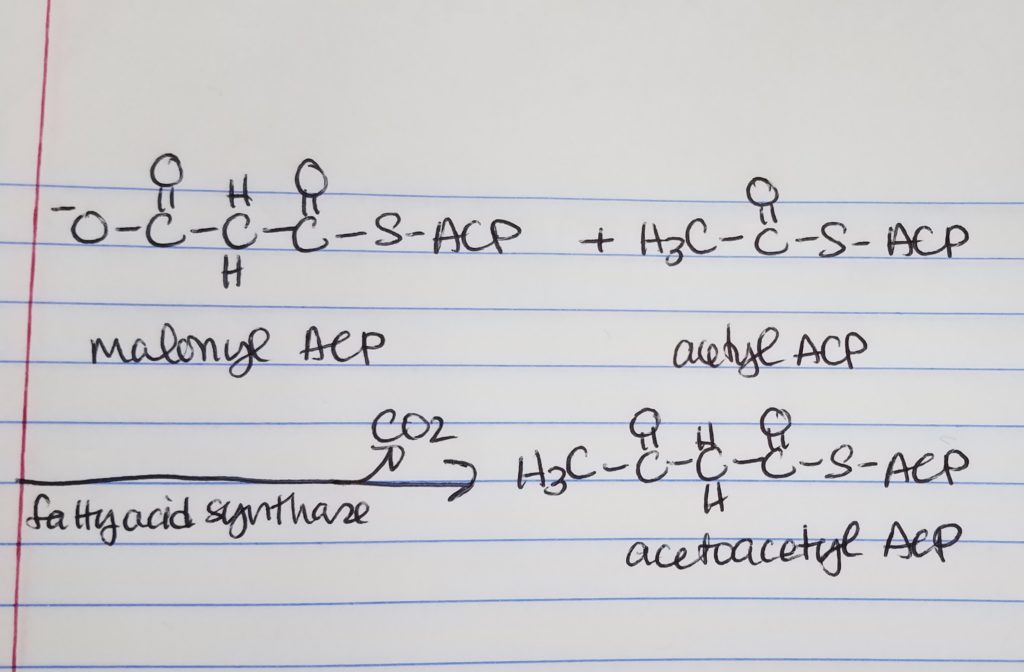
Fatty Acid Synthesis Step 2. Reduction.
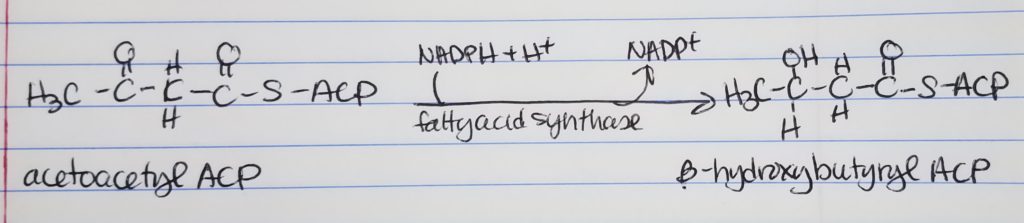
Fatty Acid Synthesis Step 3. Dehydration.
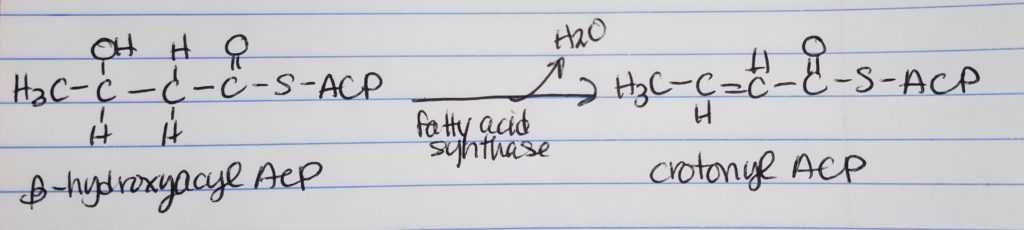
Fatty Acid Synthesis Step 4. Reduction.
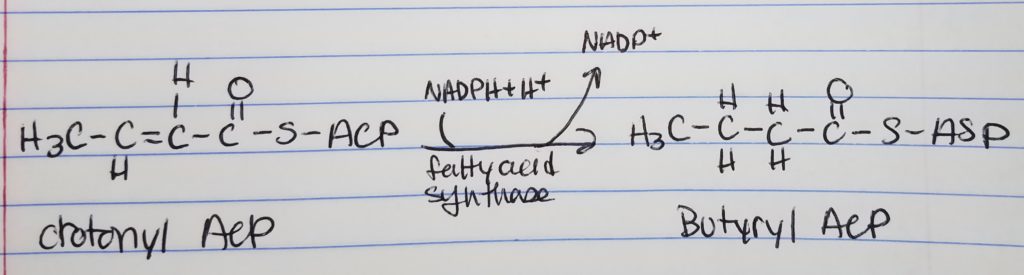
How to Make a Long Fatty Acid? Keep repeating…
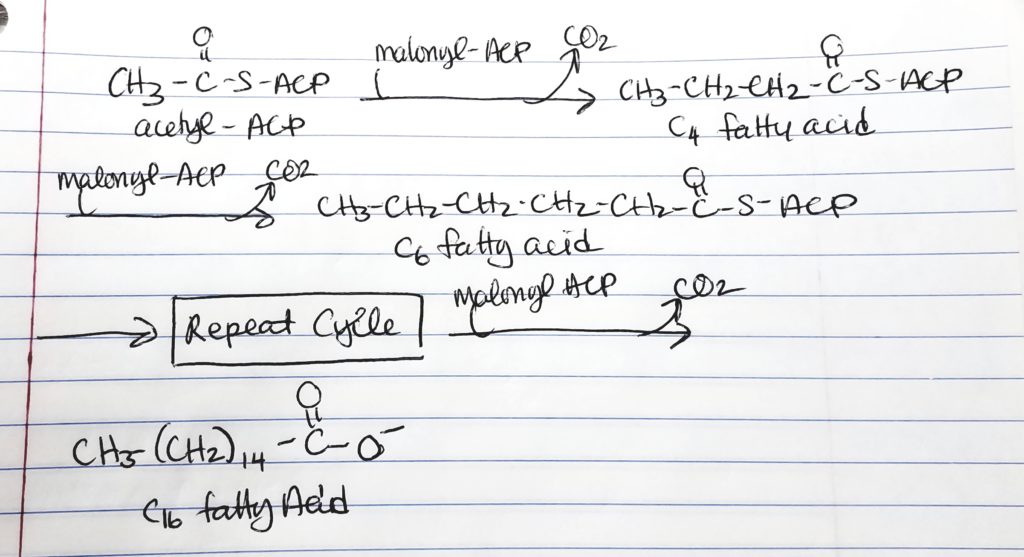
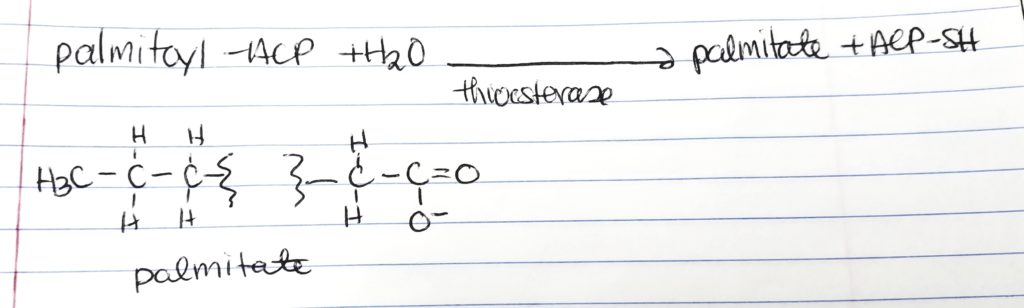
Palmitate.
Elongation and Unsaturation.
Summary of Key Points of FA Synthesis Steps.
Release Palmitate.
Elongation an Desaturation.
Regulation of Fatty Acid Synthesis.
Global Regulation.
Local Regulation.
Fed.
Fasting.
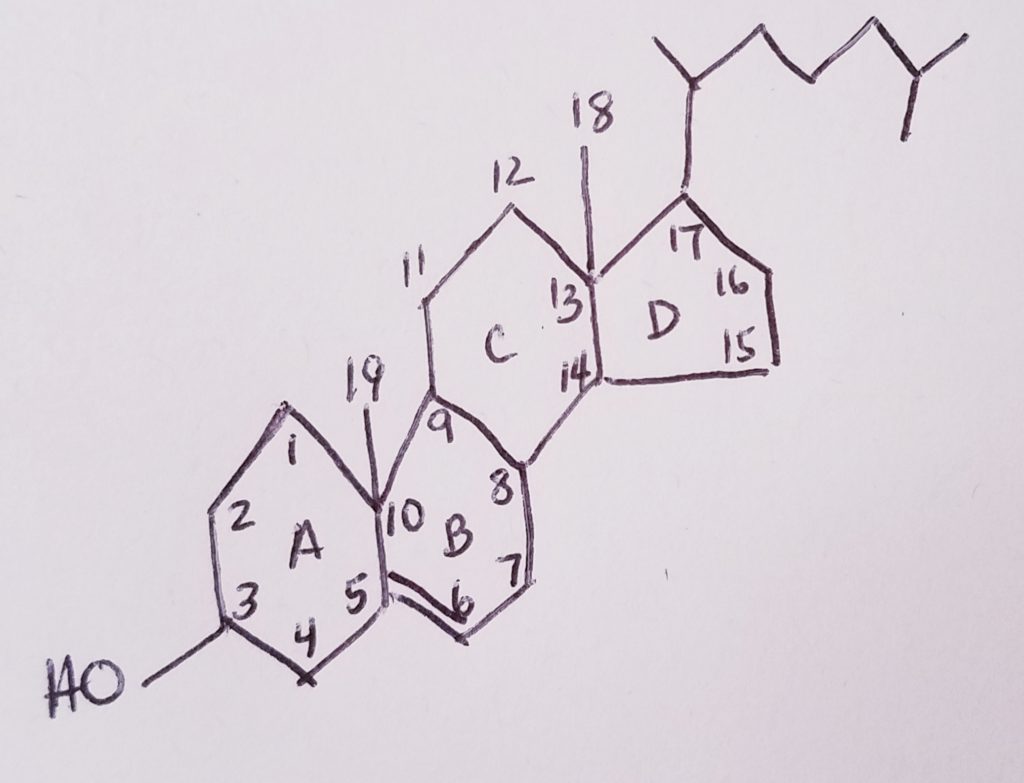
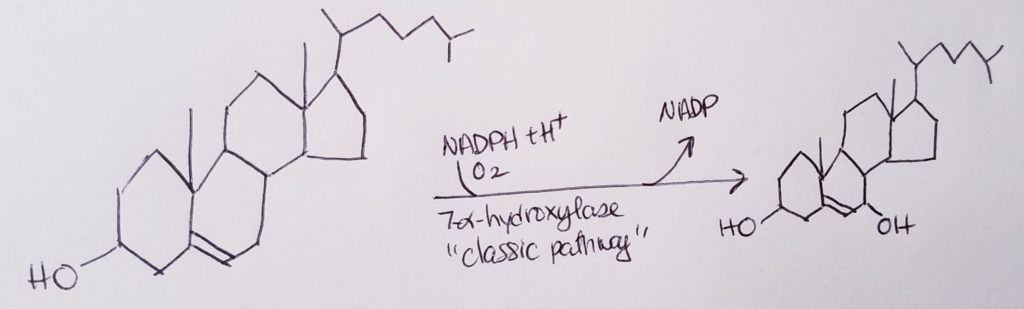
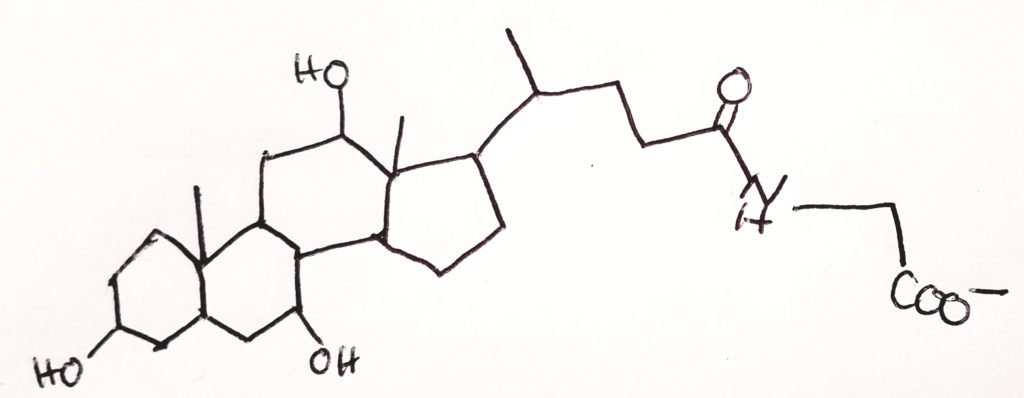
Cholesterol Metabolism.
Phytosterol.
Flow of Major Cholesterol Intermediates: Number of Carbons and Number of Acetyl CoA.
Stage 1. Acetyl CoA to Mevalonate (2-carbons to 6-carbons).
Stage 2. Mevalonate to Isopentenyl Pyrophosphate (IPP) (6-carbons, -1, 5-carbons).
Stage 3A. IPP to Farnesyl Pyrophosphate.
Stage 3B. Farnesyl Pyrophosphate to Squalene.
Stage 4. Squalene to Lanosterol (both are 30 carbons long).
Stage 5. Lanosterol to Cholesterol (30 carbons to 27 carbons).
| Hydrolyzable | Non-Hydrolyzable |
|---|---|
| Esters | Hydrocarbons |
| Fats | Carotenoids |
| Waxes | Alcohols |
| Sterol Esters | Sterols |
| Phospholipids | Steroids |
| Phosphatidates | Acids |
| Phosphtids | Fatty Acids |
| Spingolipids | Eicosanoids |
| Glycolipids |
Regulation of HMG-CoA Reductase.
Statins, Muscle Pain, Ubiquinone (CoQ).
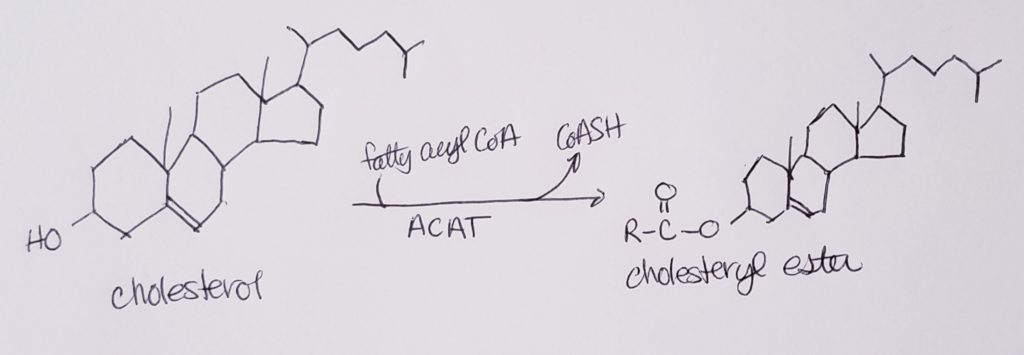
Cholesterol Esterification.
Bile Acids and Bile Salts.
Vitamin D, Calcitriol.
5 Types of Steroid Hormones Derived from Cholesterol.
Lipoproteins (transport cholesterol).
Apolipoproteins (bind lipids to proteins to make lipoprotein).
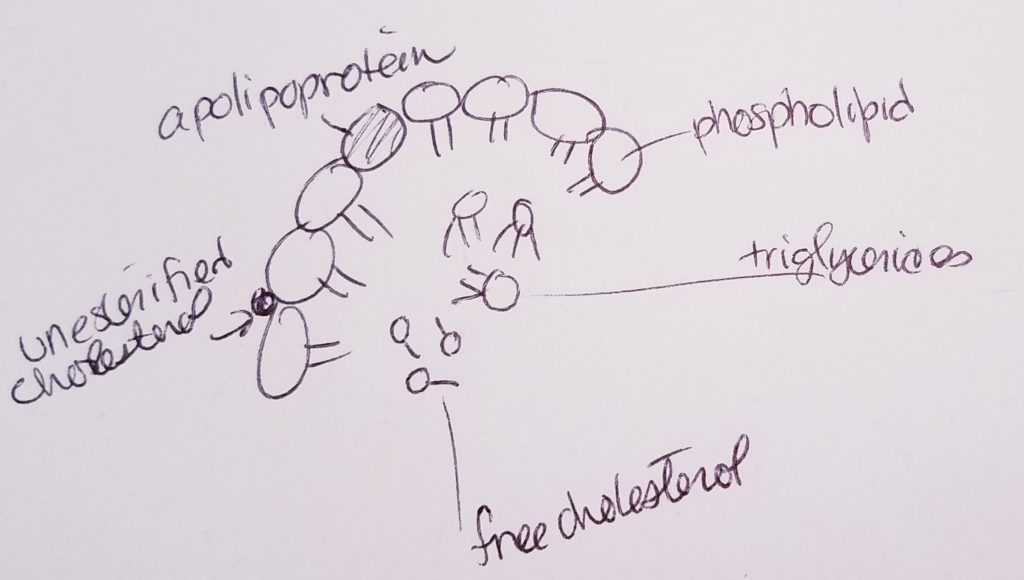
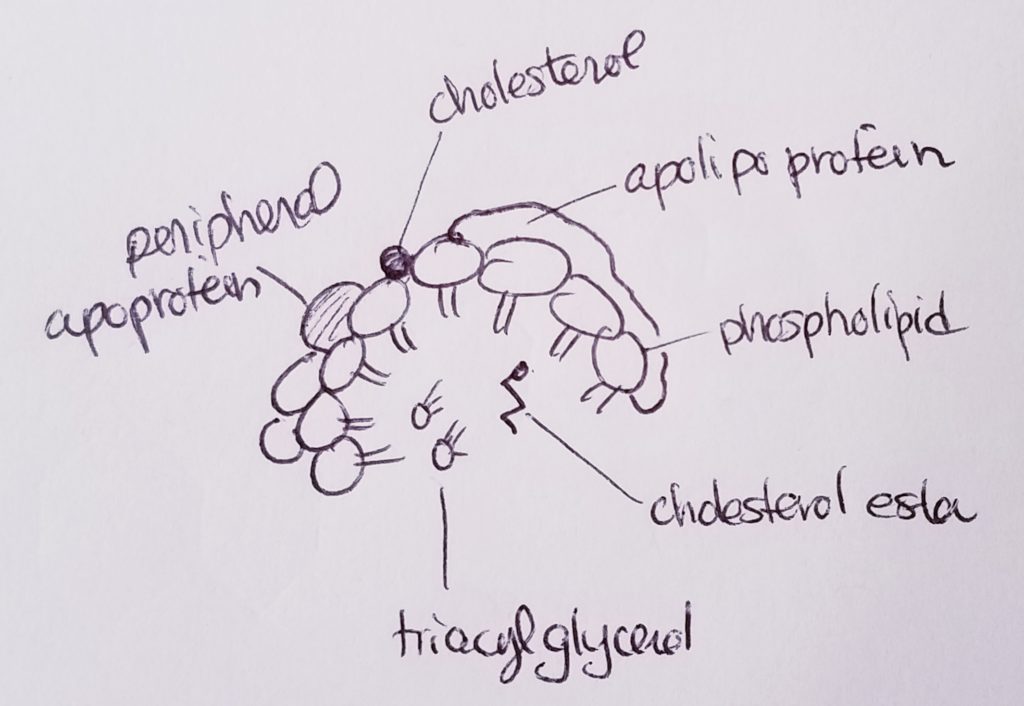
Hormones.
Fed & Fasting States.
Insulin.
Regulators of Insulin.
Regulators of Glucagon.
Other Regulators.
Thought Exercise: What Happens Right After a Meal vs. What Happens a Few Hours After a Meal??
Digestion.
Digestive Path.
Resources.
References.
Bean, J. (2019). Cholesterol metabolism.
Bean, J. (2019). Fatty acid synthesis.
Bean, J. (2019). Fed and fasting states.
Ferrier, D. (2017). Biochemistry (7th ed.). Philidelphia, PA: Lippincott Illustrated Reviews.
Lieberman, M., & Peet, A. (2017). Marks’ basic medical biochemistry: A clinical approach(5th ed.). Philadelphia, PA: LWW.
Last edited: 08.24 .2019
Outline.
Glycogen.
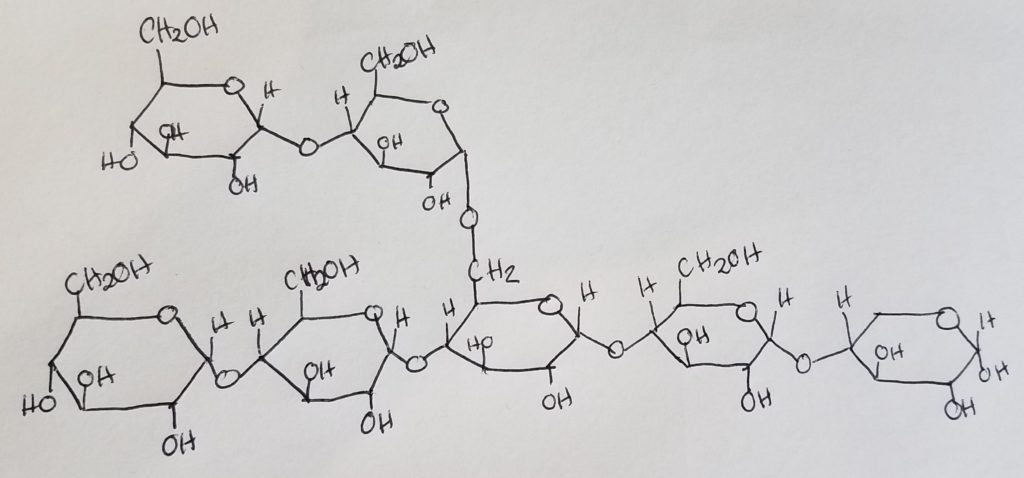
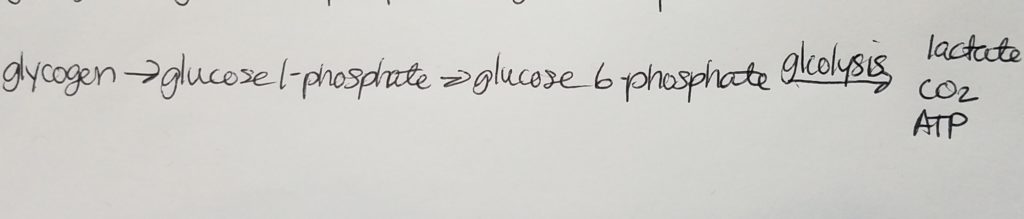

Glycogenesis (Glycogen synthesis).
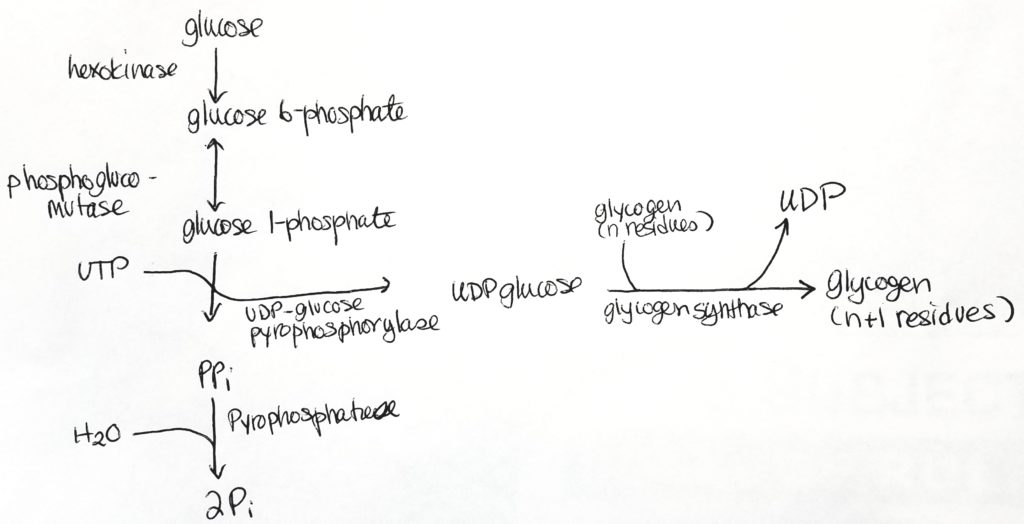
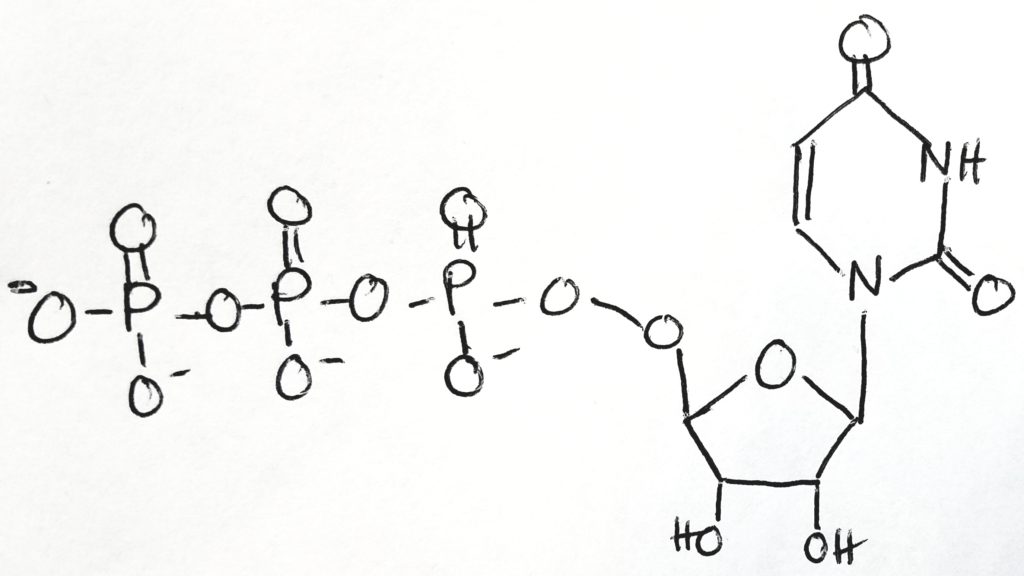
Uridine-5′-triphosphate ( C9H15N2O15P3 ).
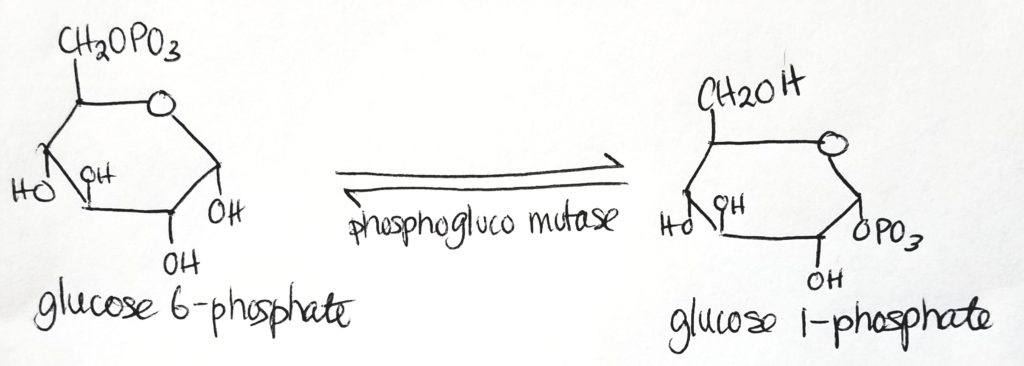
Step 1.
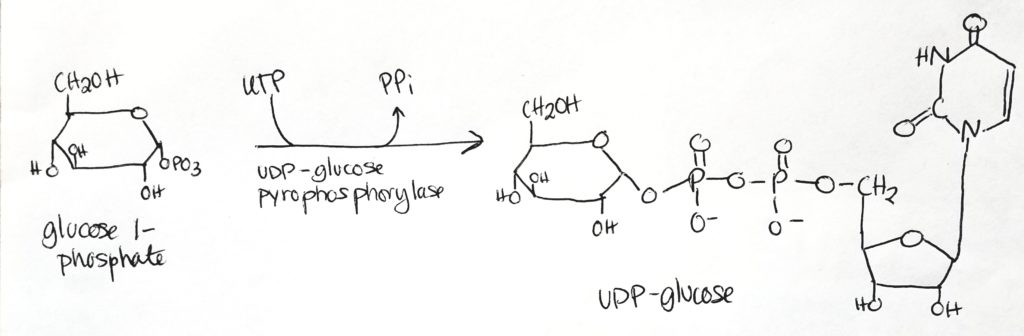
Step 2.
Step 3.
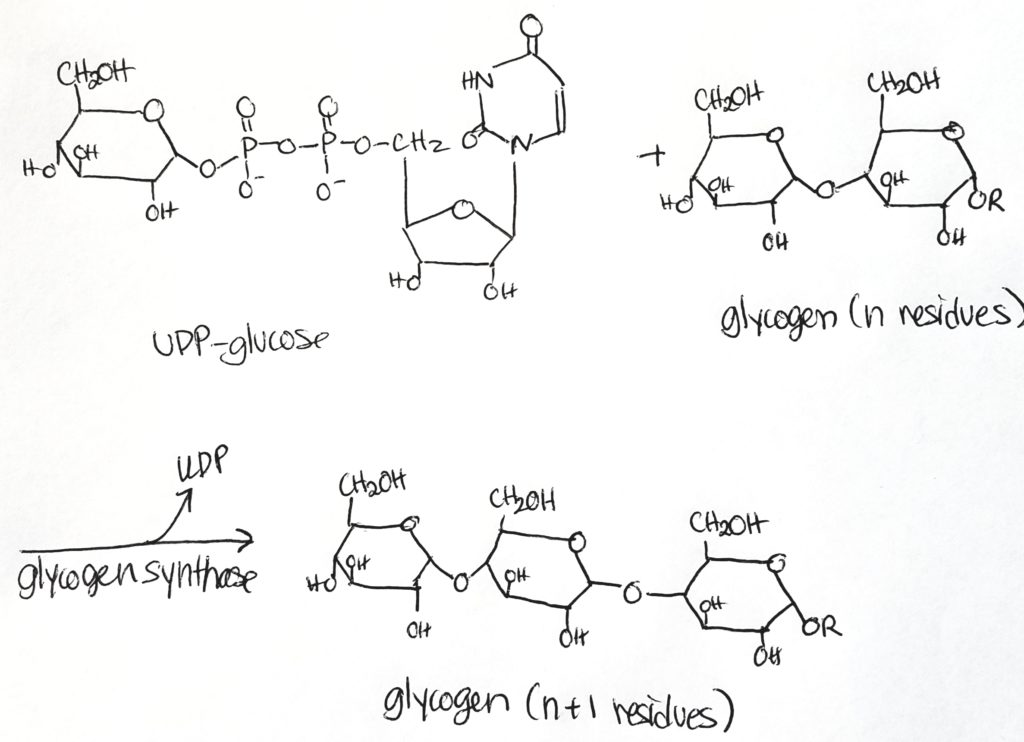
Glycogen Synthesis Enzymes.
Glycogenolysis.
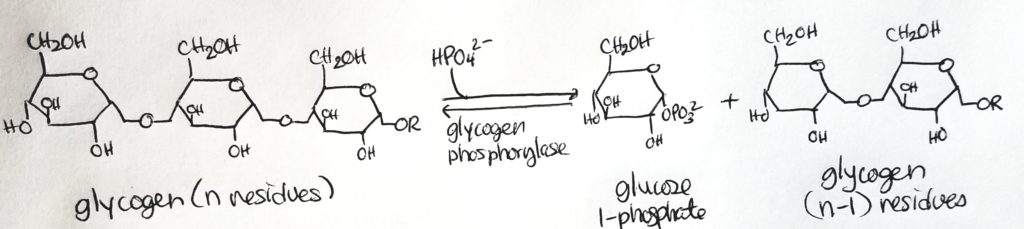
Glycogen Debranching Enzyme.
Regulation of Glycogen Metabolism.
Insulin.
Glucagon and Epinephrine.
Fatty Acid Metabolism (Oxidation).
Fatty Acid Breakdown.
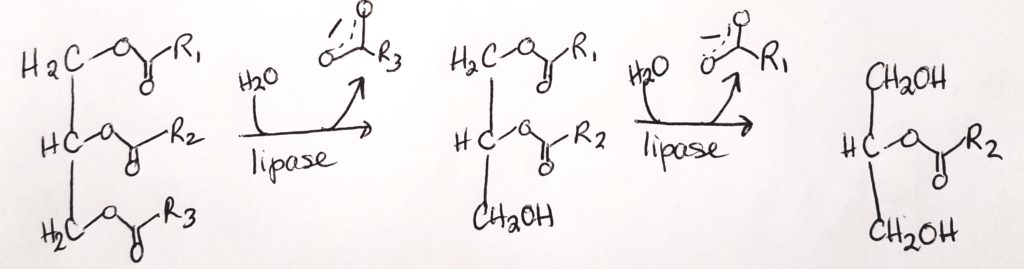
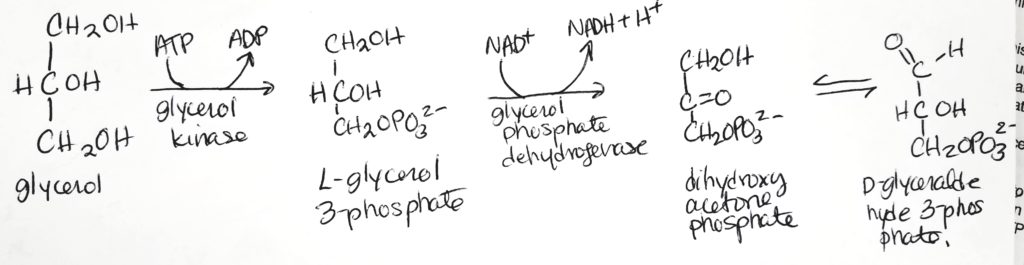
Triglyceride Digestion & Absorption.
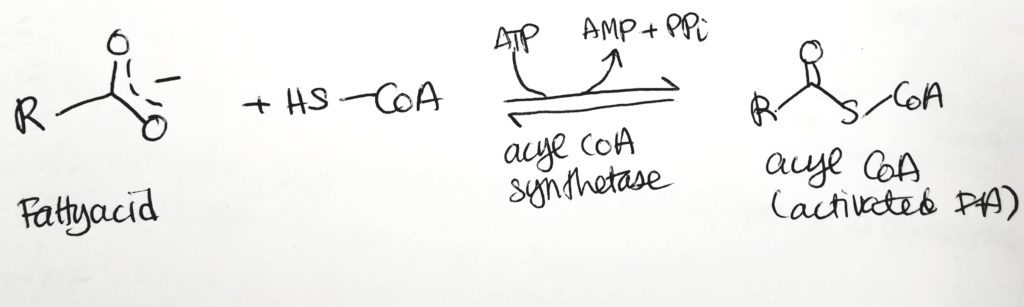
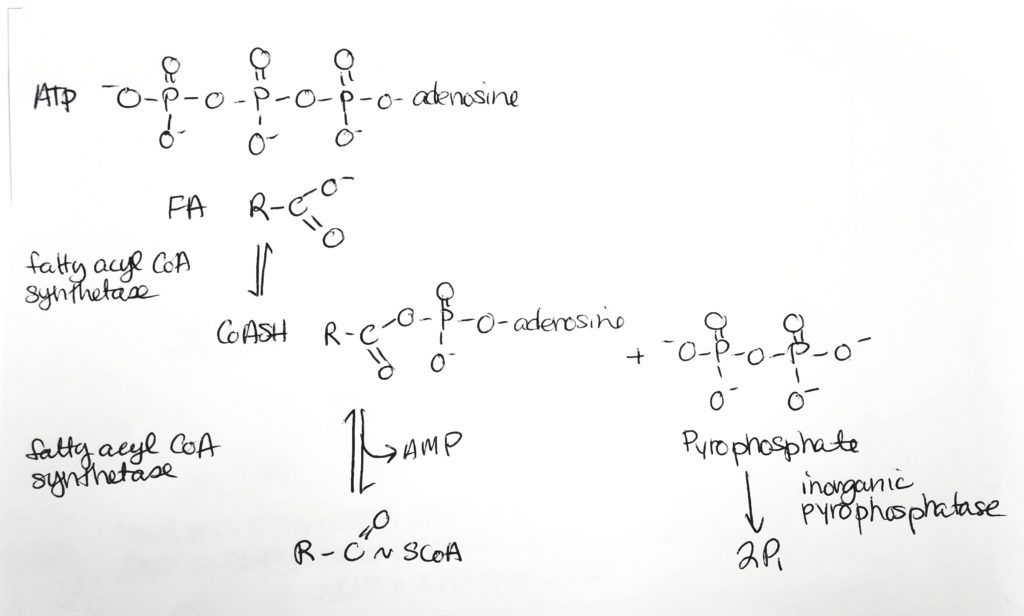
Fatty Acid Activation.
Getting the Fatty Acid into the Matrix.
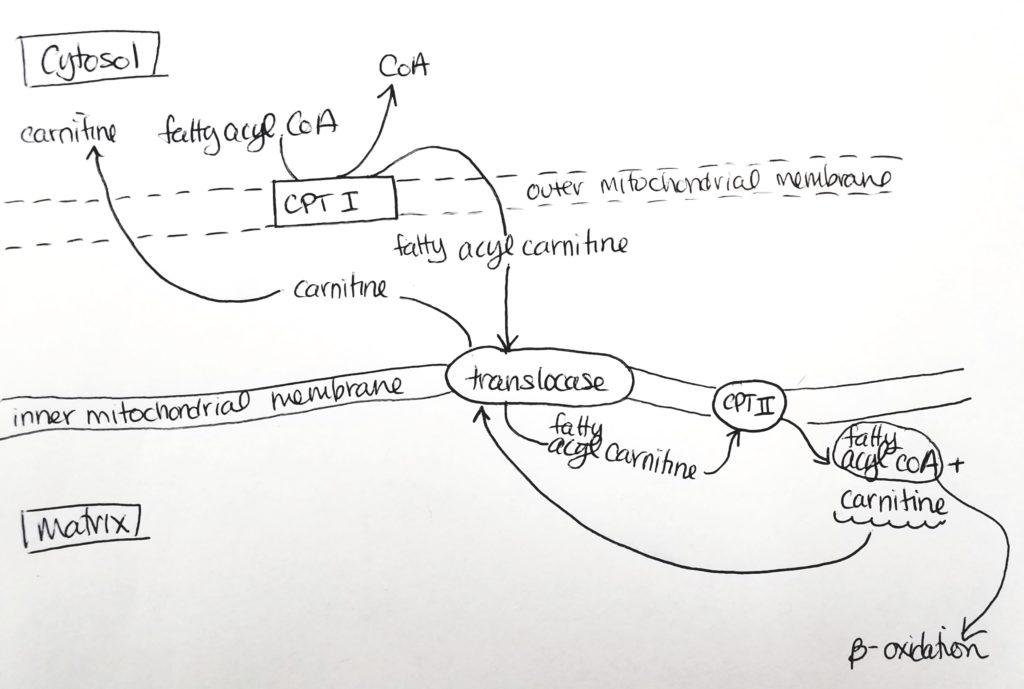
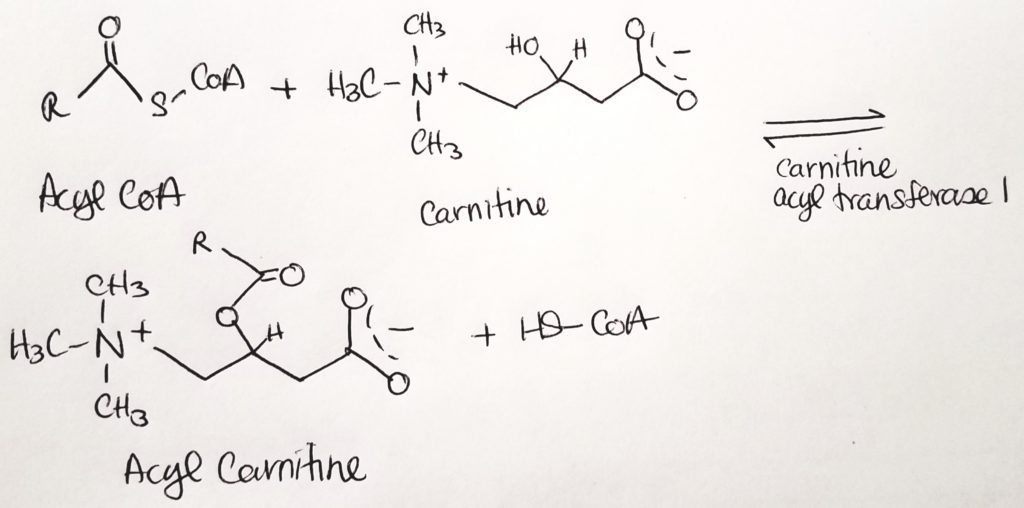
Beta-Oxidation.
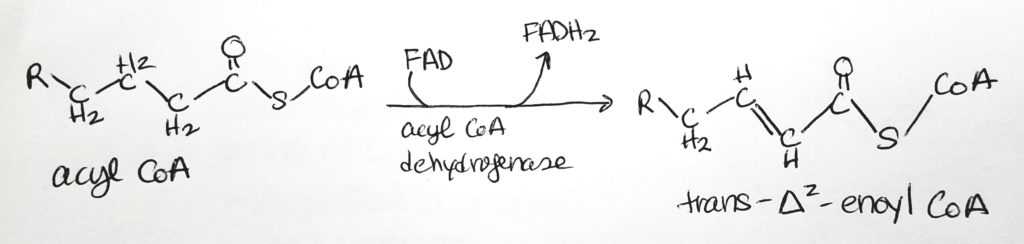
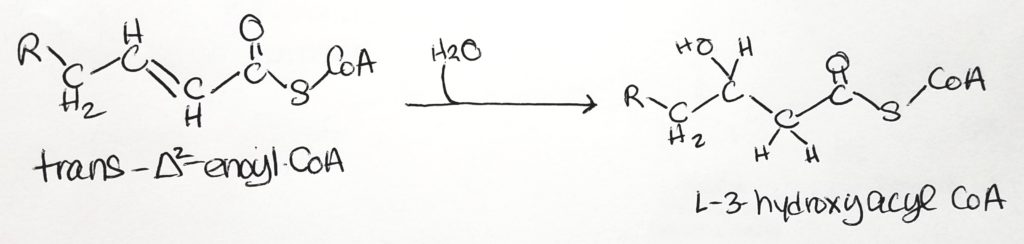
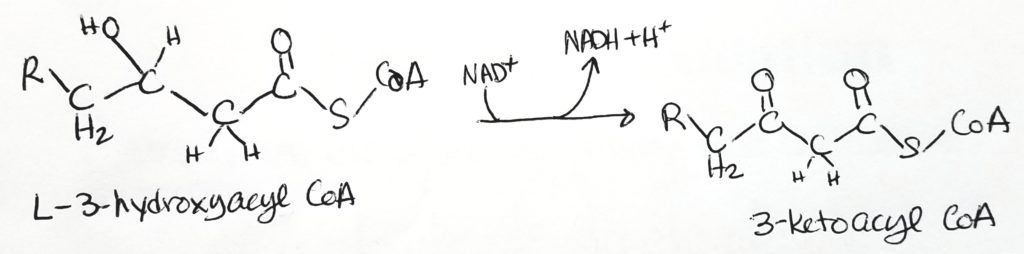
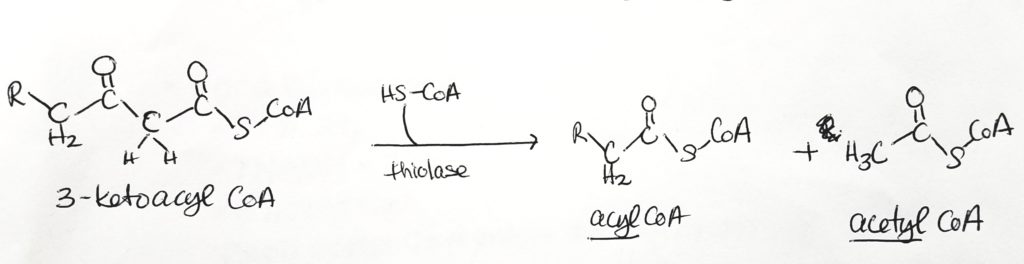
Calculating Energy Exchange of Fatty Acid Oxidation. What do you really get?
What do you do with an odd-numbered FA chain?
Don’t panic. Odd numbered FA’s make propionyl-CoA. The last fragment of 3 carbons is called propionyl-CoA. HCO3- (bicarb) and ATP are added (coenzyme biotin). Coenzyme B12 is also active to make 4-carbon succinyl-CoA.
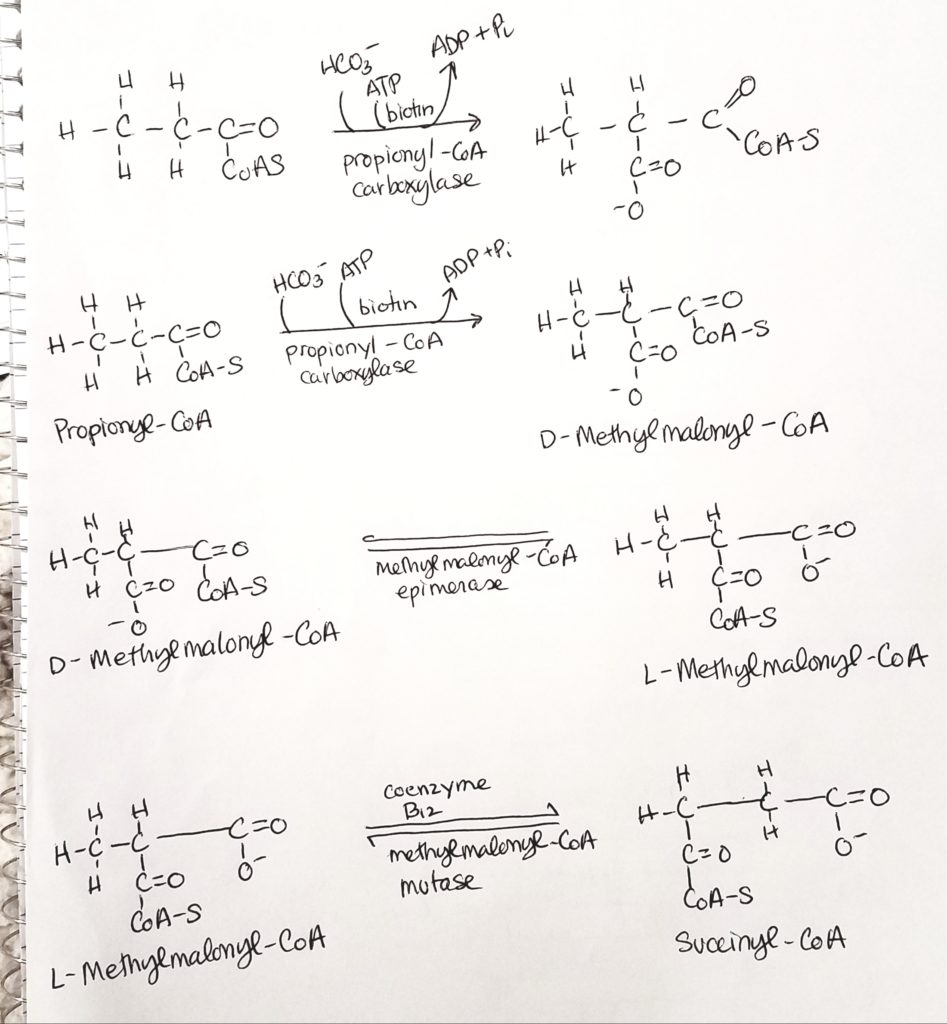
Let’s work an odd-numbered problem.
Fatty Acid Oxidation: Animals vs. Plants.
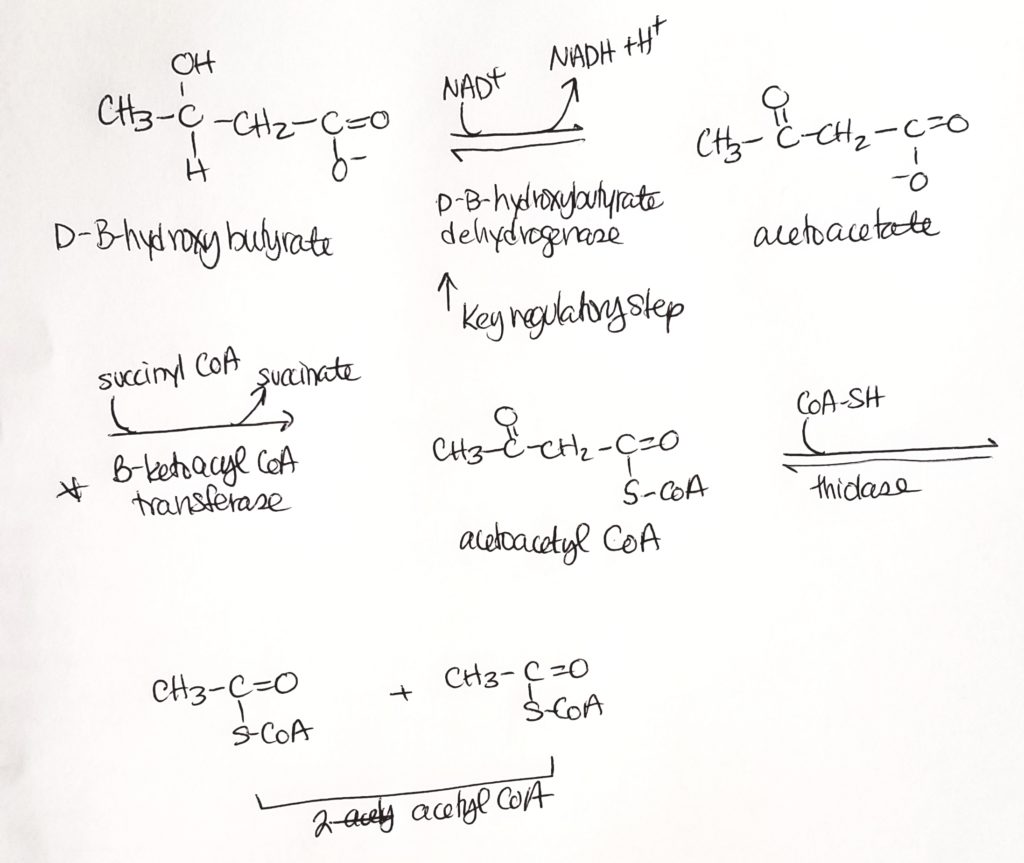
Ketone Body Metabolism.
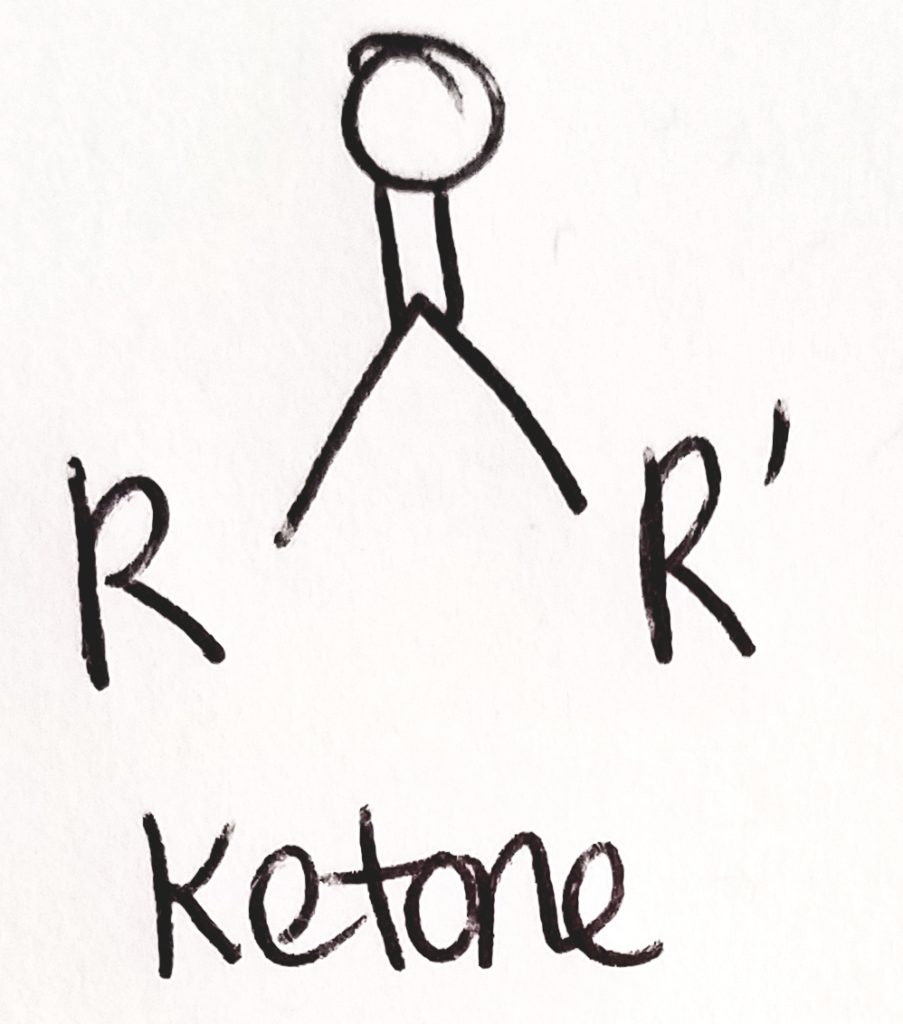
What are ketone bodies?
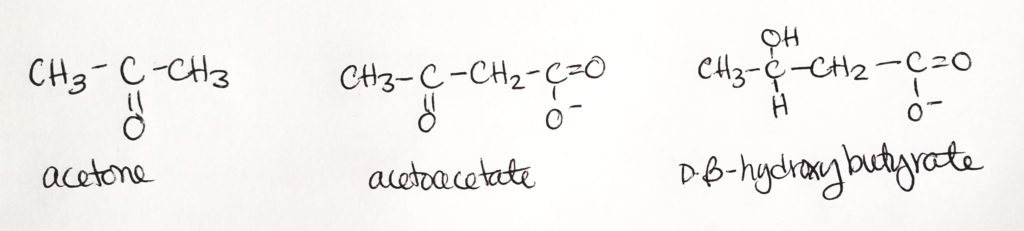
Synthesis of Ketone Bodies.
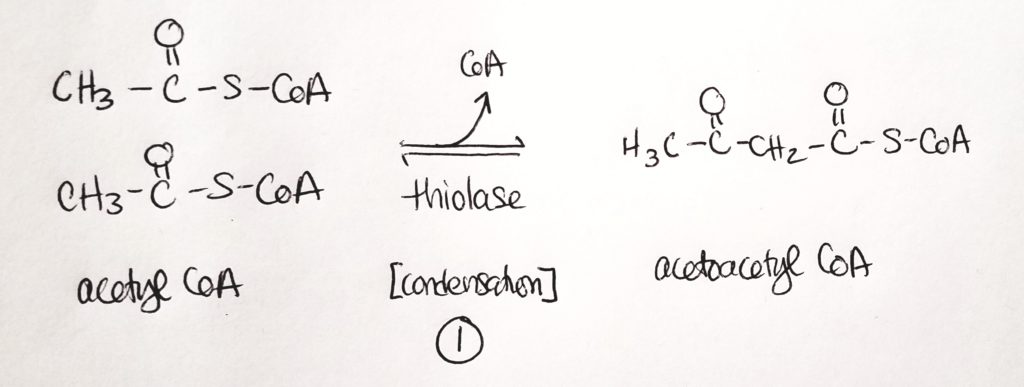
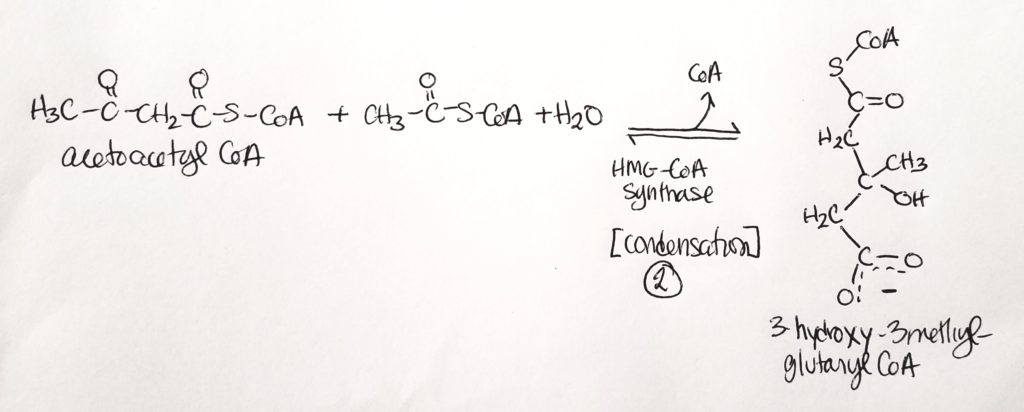
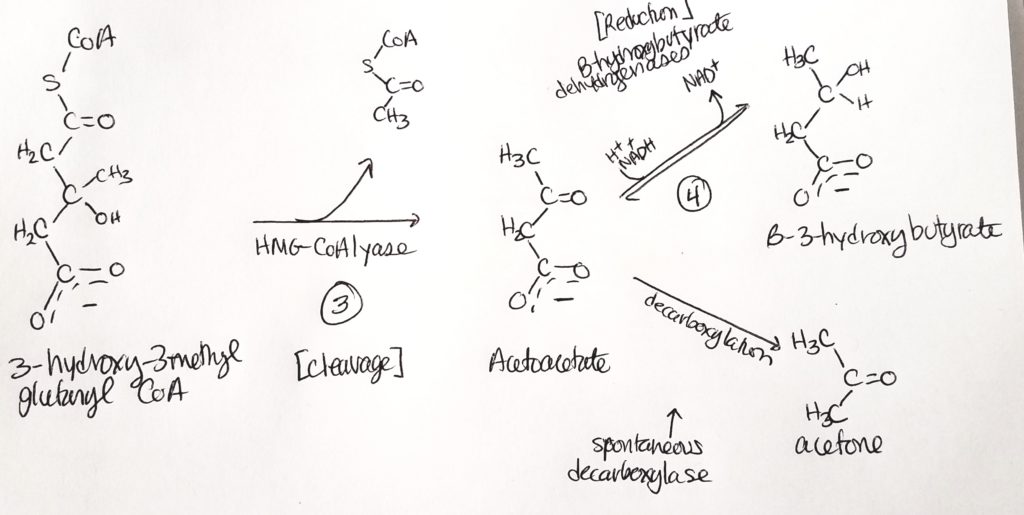
Ketone Body Oxidation (we made them, now we break them).
Amino Acid & Nitrogen Metabolism.
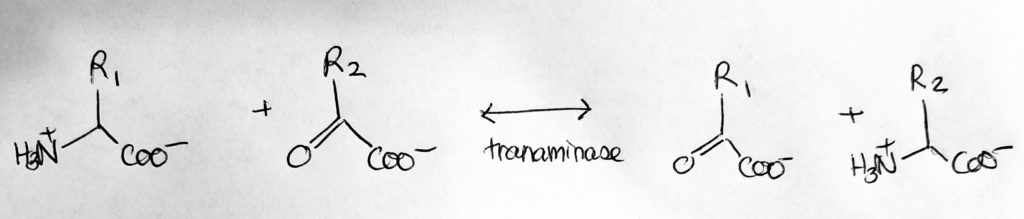
How to move Nitrogen around: Transamination.
How to move Nitrogen around: Amination.

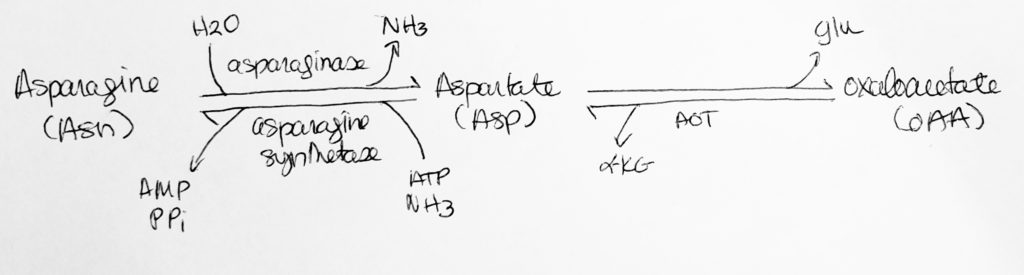
How to move Nitrogen around: Deamination.
Fate of AA Carbons and Nitrogen.
Glucogenic & Ketogenic AA Degradation.
Urea (ornithine) Cycle.
Resources.
References.
Bean, J. (2019). Amino acid nitrogen metabolism.
Bean, J. (2019). Fatty acid metaboslim.
Bean, J. (2019). Glycogen metaboslim.
Bean, J. (2019). Ketone body metabolism.
Ferrier, D. (2017). Biochemistry (7th ed.). Philidelphia, PA: Lippincott Illustrated Reviews.
Lieberman, M., & Peet, A. (2017). Marks’ basic medical biochemistry: A clinical approach(5th ed.). Philadelphia, PA: LWW.
Ferrier, D. (2017). Biochemistry (7th ed.). Philidelphia, PA: Lippincott Illustrated Reviews.
Lieberman, M., & Peet, A. (2017). Marks’ basic medical biochemistry: A clinical approach(5th ed.). Philadelphia, PA: LWW.
Last edited: 08.14.2019
Outline.
TCA Cycle.
Net Equation.
3NAD+ + FAD + FDP + Pi + Acetyl-CoA + 2H2O >>> 3NADH + FAD(2H) + GTP + CoASH + 2CO2
Quick Redox Review.
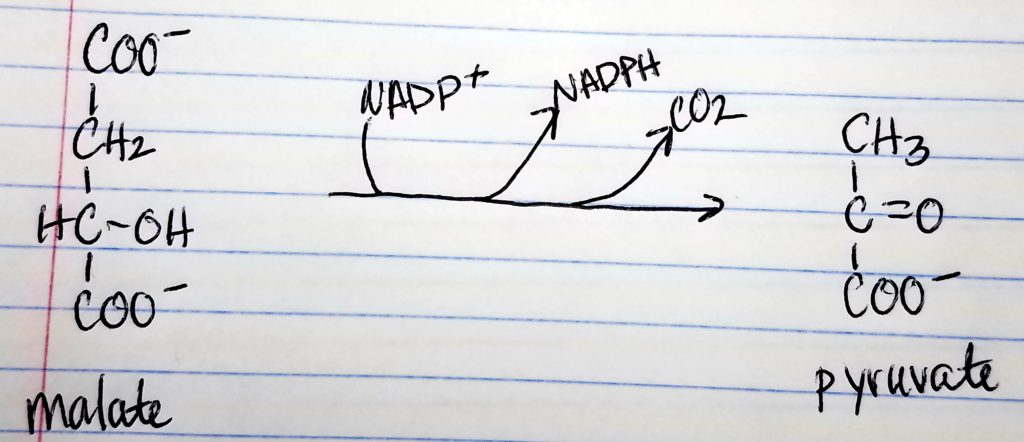
Acetyl CoA.
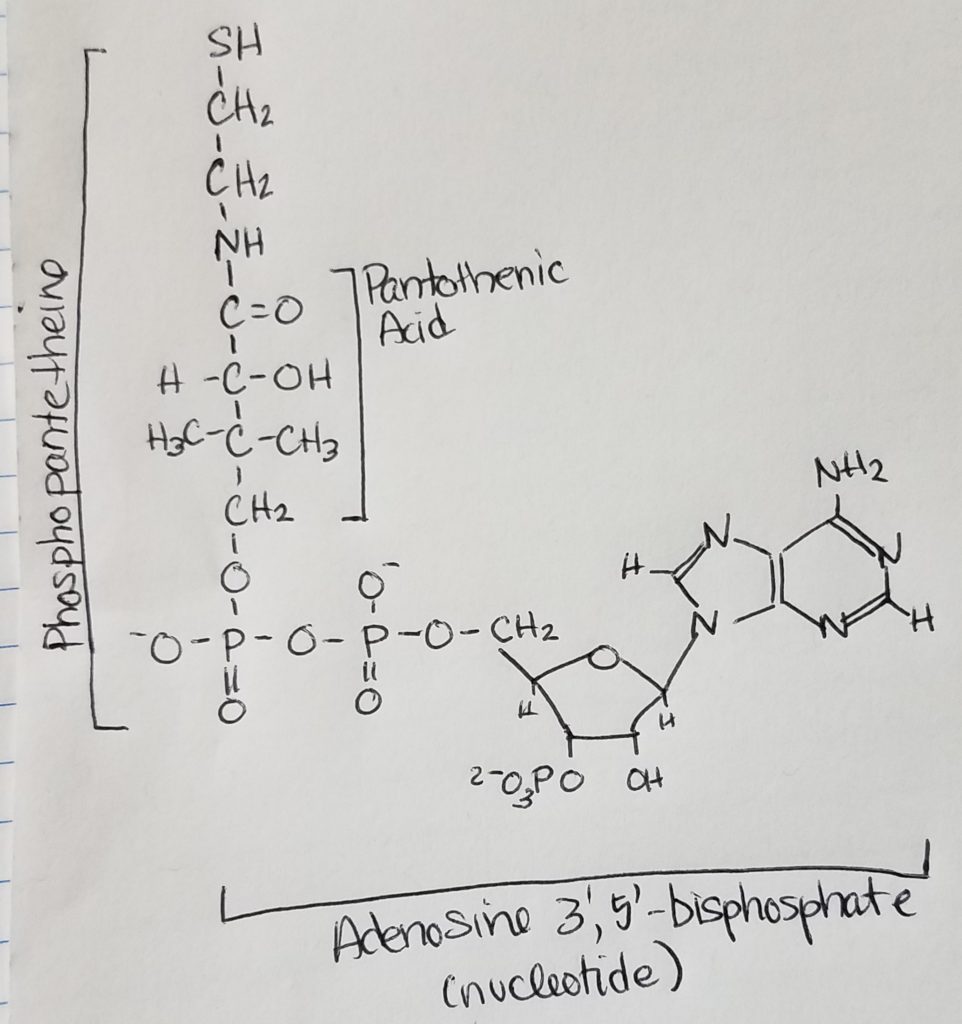
Pyruvate Dehydrogenase Complex (PDHC/PDC).
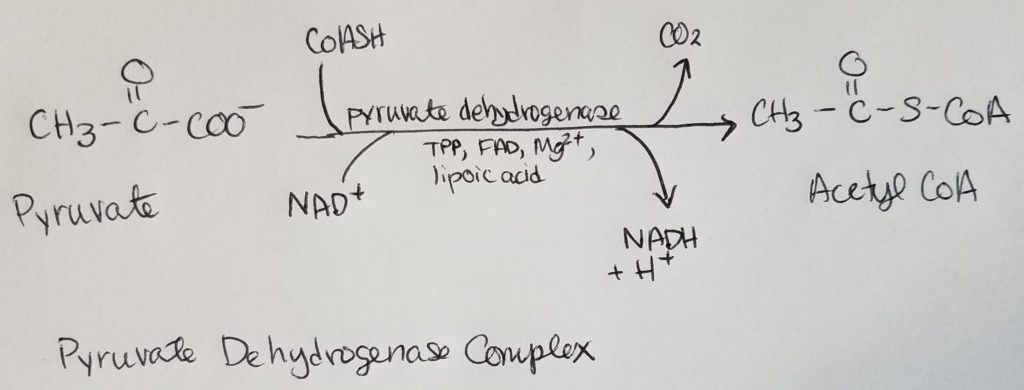
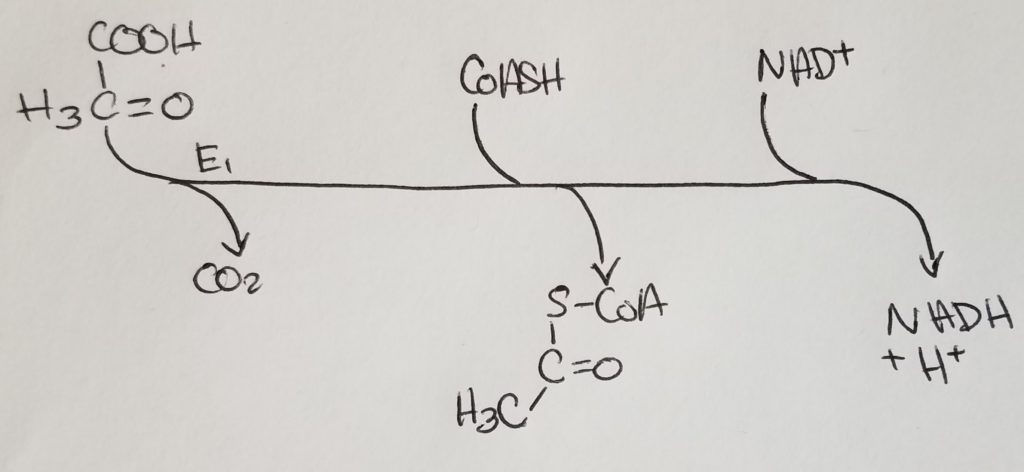
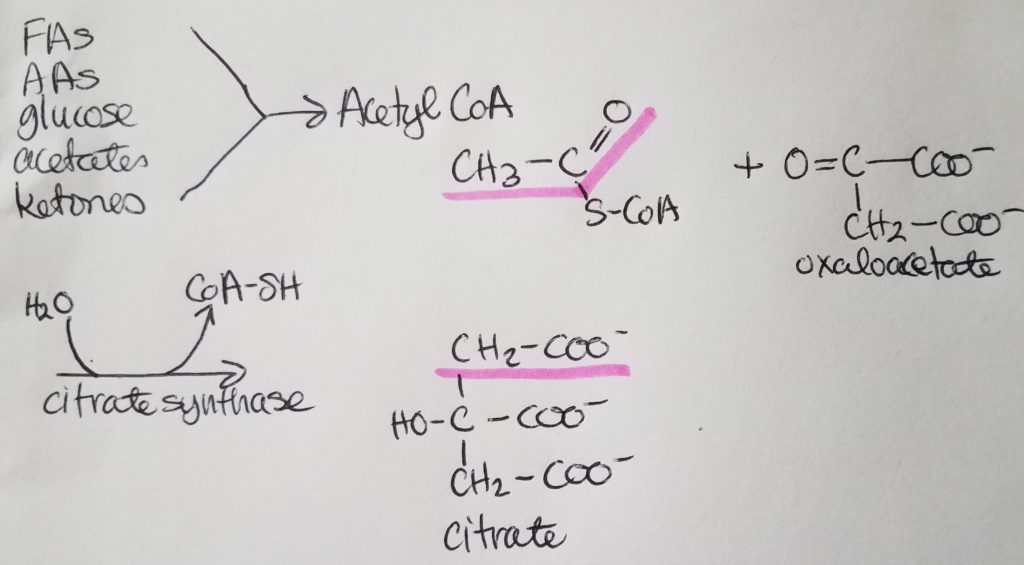
STEP 1. Citrate Synthase Rxn.
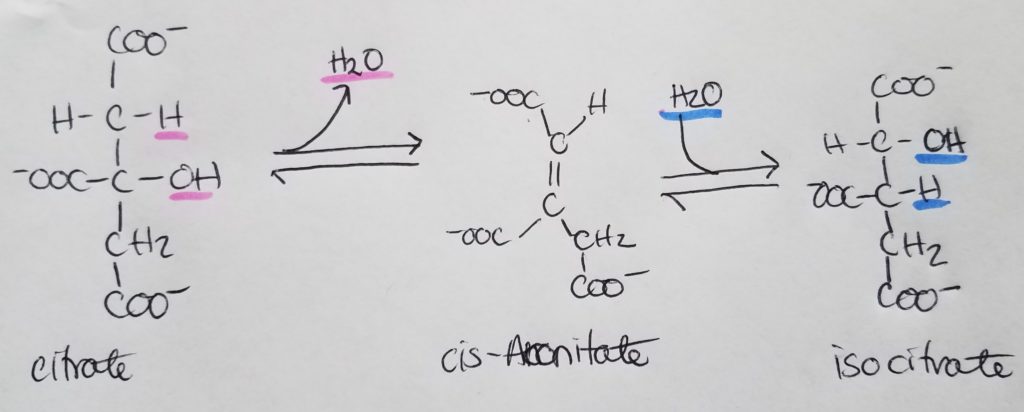
STEP 2. Isomerization of Citrate.
STEP 3. Isocitrate Dehydrogenase Rxn.
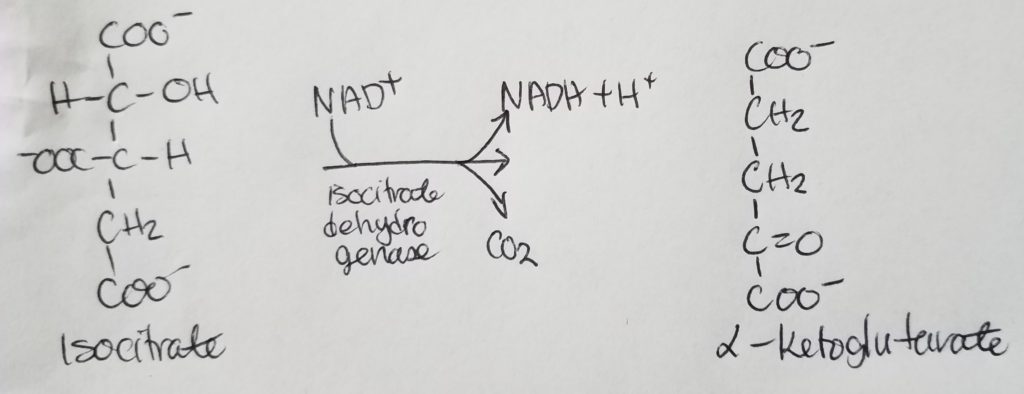
STEP 4. Oxidation of Alpha-Ketoglutarate.
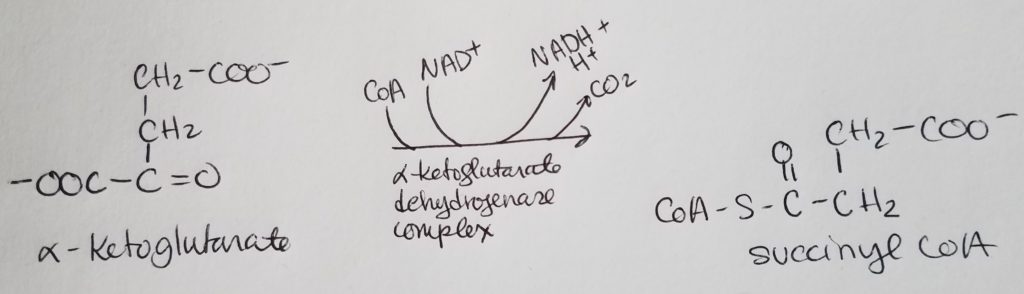
STEP 5. Formation of Succinate.
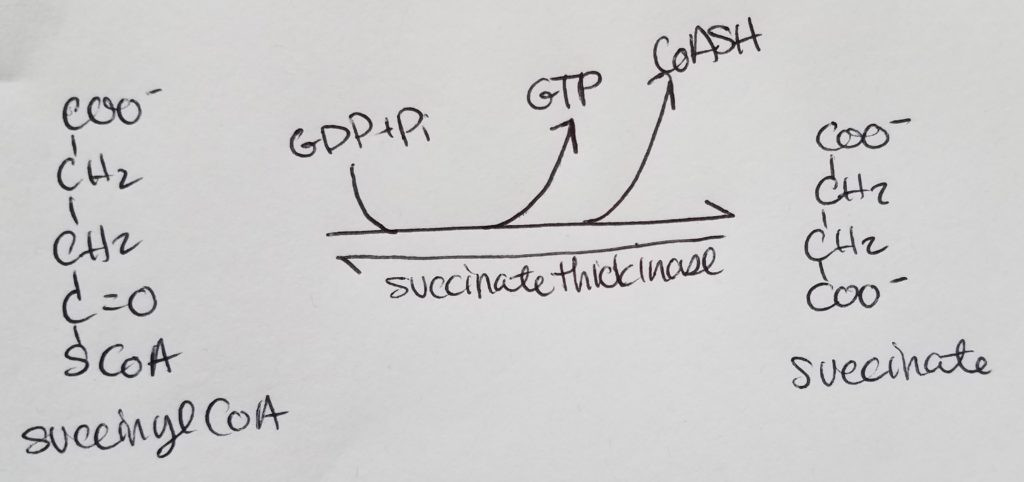
STEP 6. Formation of Fumarate.
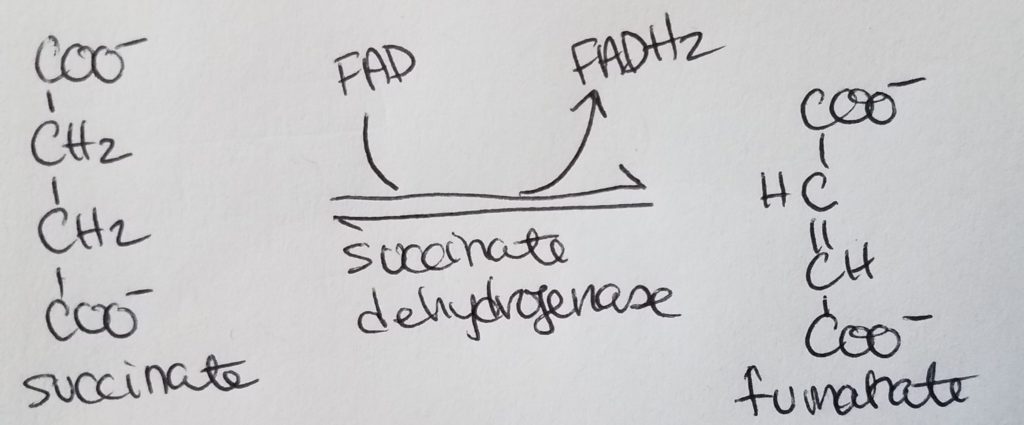
STEP 7. Formation of Malate.
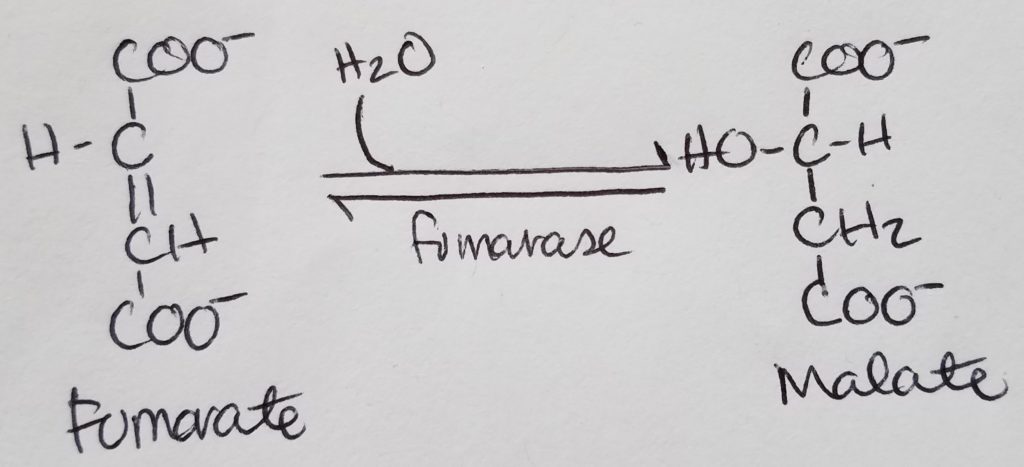
STEP 8. Formation of Oxaloacetate.
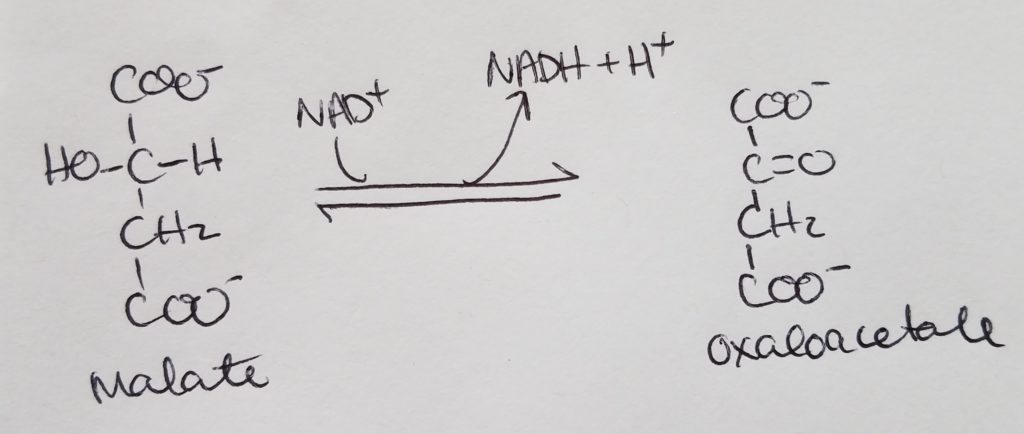
Summary of TCA Step 1-8.

TCA Regulation.
Citrate Synthase Regulation.
Isocitrate Dehydrogenase Regulation.
Alpha-Ketoglutarate Dehydrogenase Regulation.
TCA Intermediates Regulation.
The Electron Transport Chain (ETC) & Oxidative Phosphorylation.
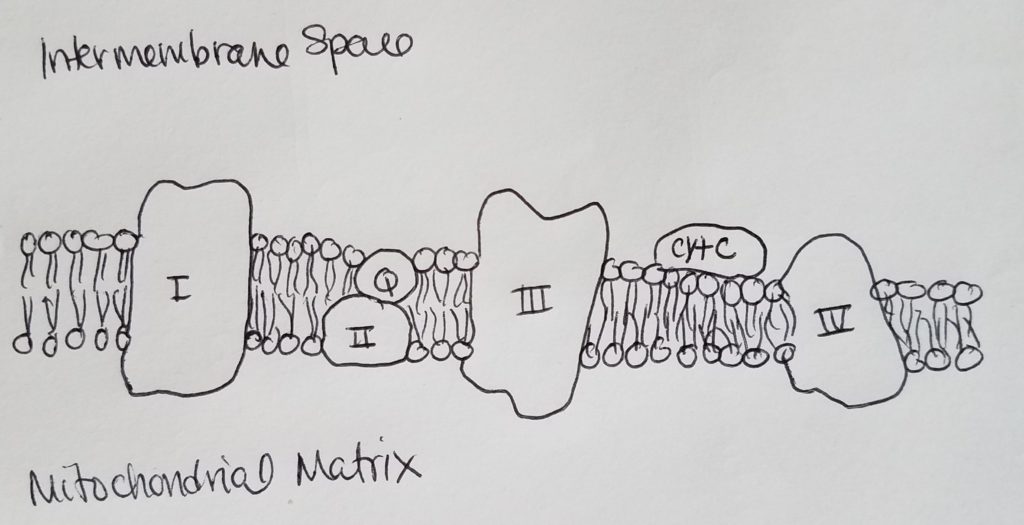
Mitochondria.
Electron Carriers.
Complex I and Q.
Complex II (succinate dehydrogenase).
Complex III (cytochrome oxidoreductase).
Complex IV.
Summary of ETC.
So thus far, the main purpose of ETC is two-fold: maintain the [H+] concentration gradient (high [H+] in the intermembrane space, and low [H+] in the matrix), and the shuttling of e- from a higher energy state to lower energy state where oxygen is the final electron acceptor and is expelled by binding it to two H+ to form water. Complexes I, III, and IV are proton pumps while complex II is like a proton funnel.
Chemiosmosis & ATP Synthase.
Glycolysis.


Stage 1. Step 1. Glucose to Glucose 6-phosphate.
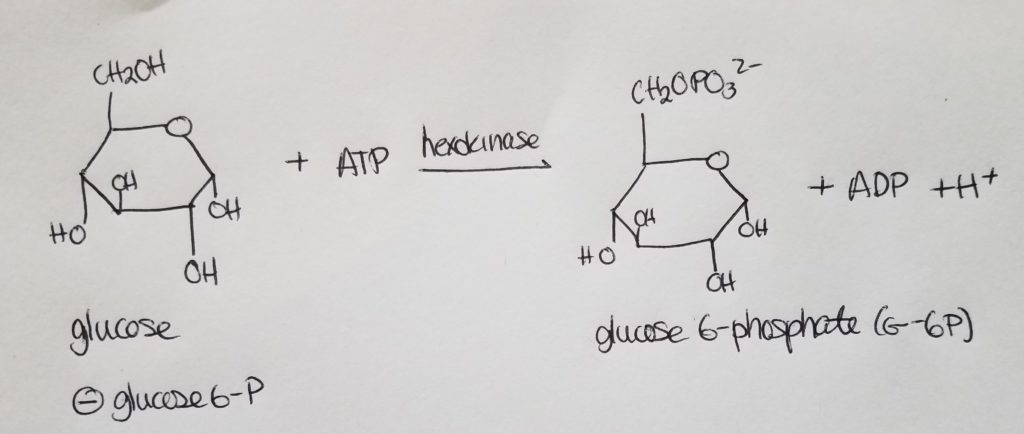
Stage 1. Step 2.
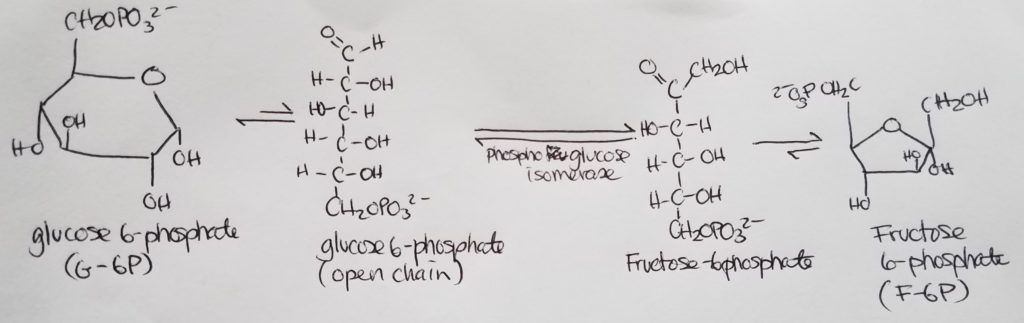
Stage 1. Step 3.
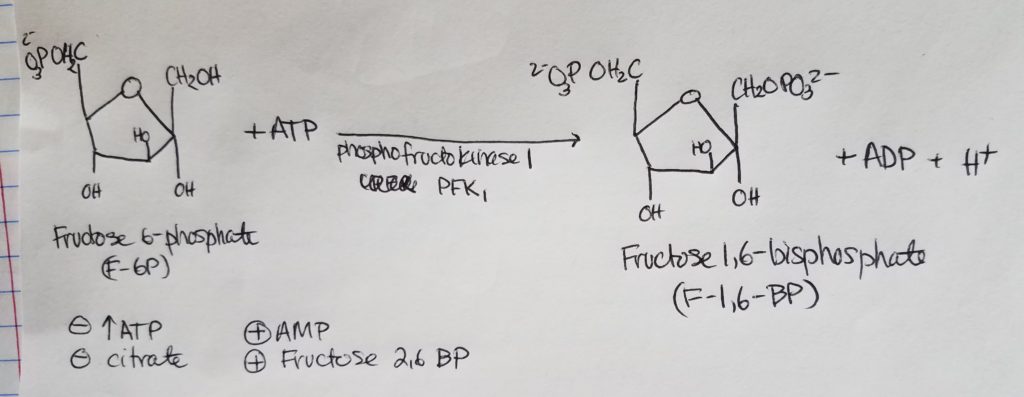
Stage 1. Step 4.
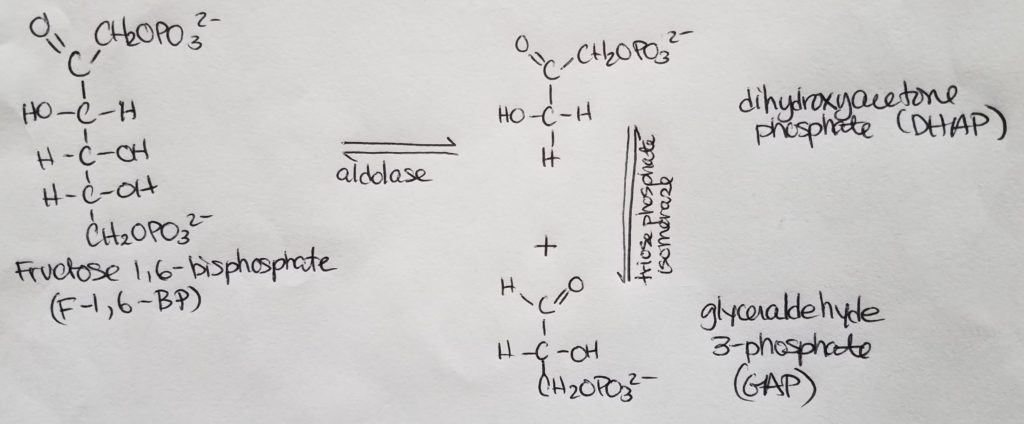
Stage 1. Step 5.
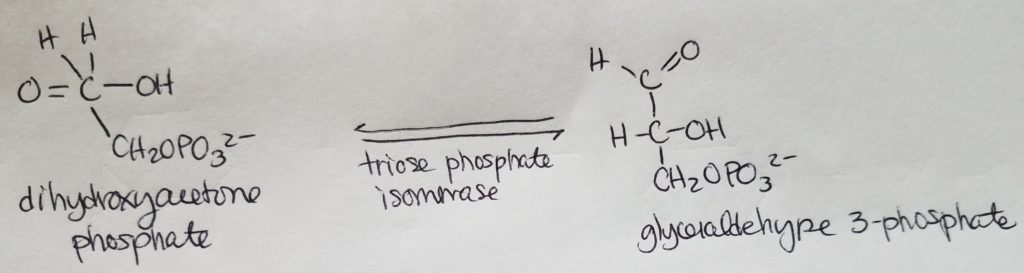
Stage 2. Step 6.

Stage 2. Step 7.
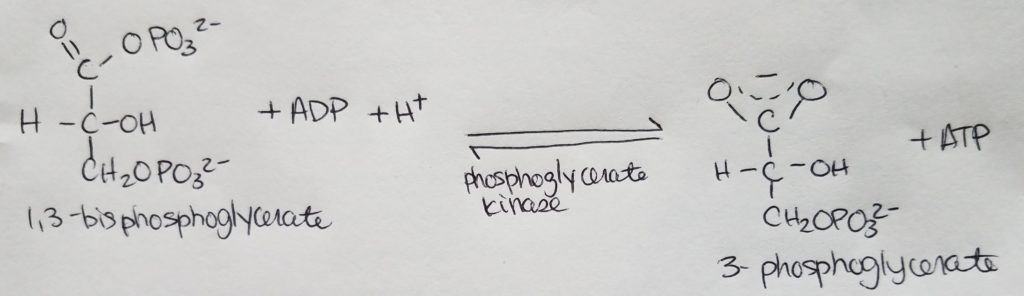
Stage 2. Step 8.
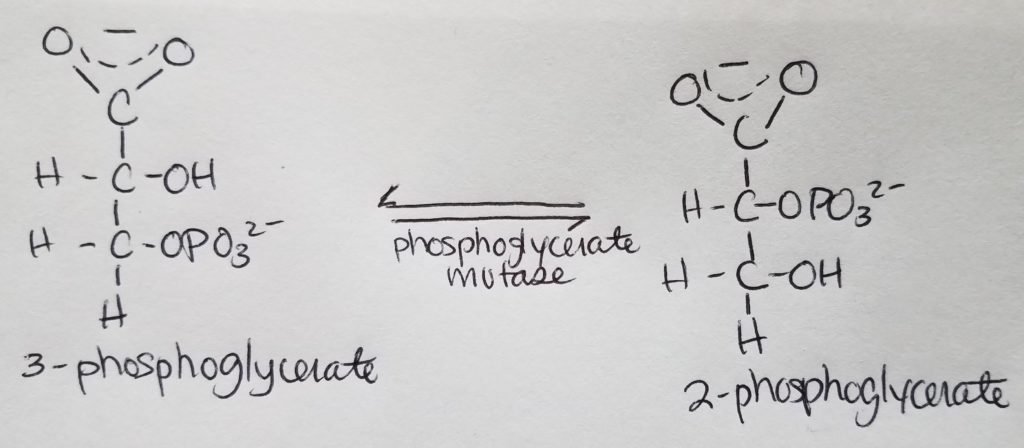
Stage 2. Step 9.
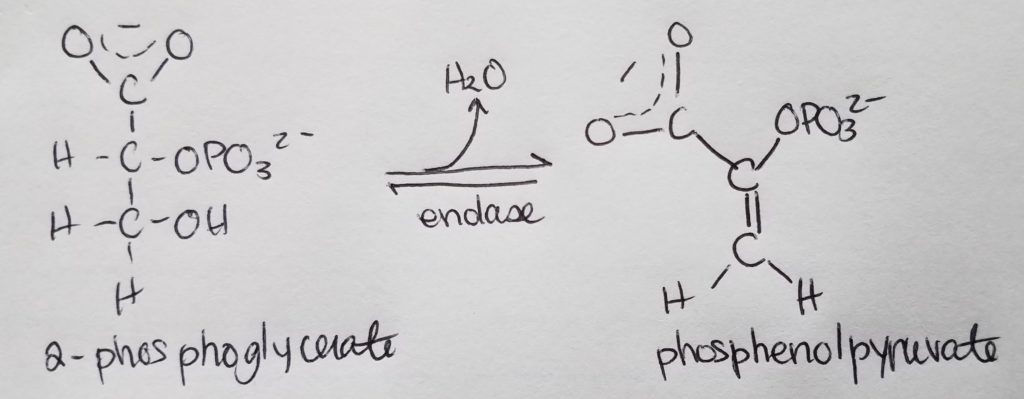
Stage 2. Step 10.
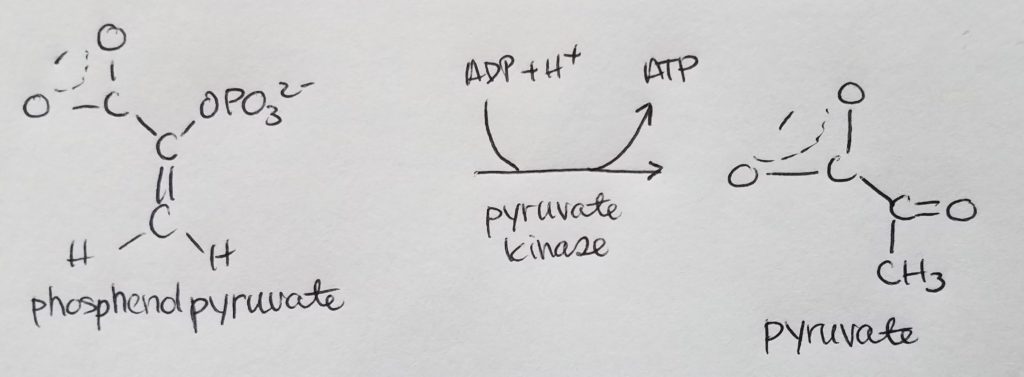
Recap.
Regulation of Glycolysis.
Alternate Paths for Pyruvate.
Alcohol Fermentation.
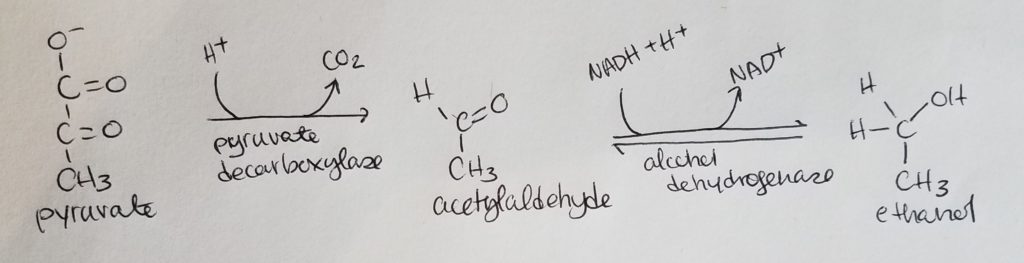
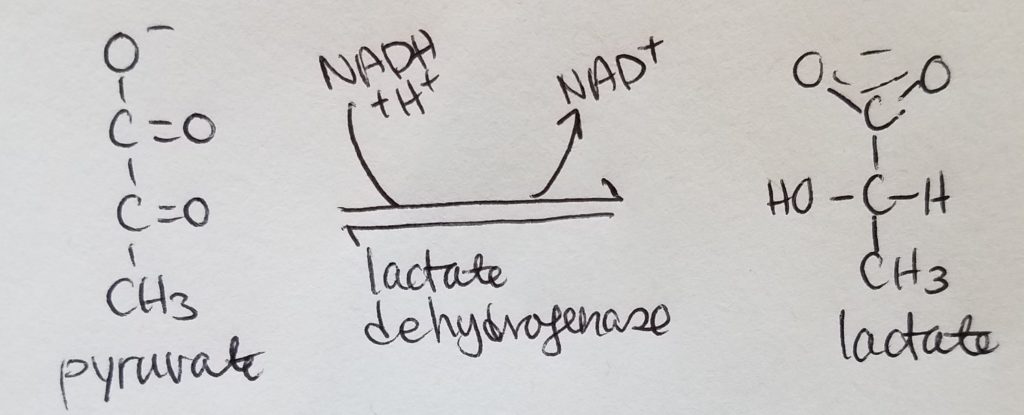
Lactate Fermentation.
Entrypoints of Other Sugars.
Gluconeogenesis.
Roadblock 1. How can we get to phosphoenol pyruvate from pyruvate?

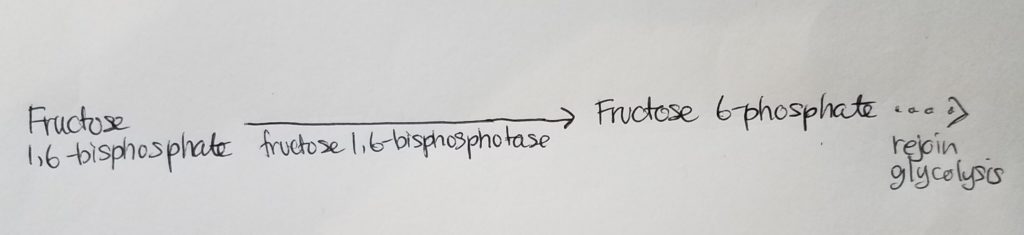
Roadblock 2. How can we get from fructose 1,6-bisphosphate to fructose 6-phosphate?
Roadblock 3. How can we get from glucose 6-phosphate back to glucose?
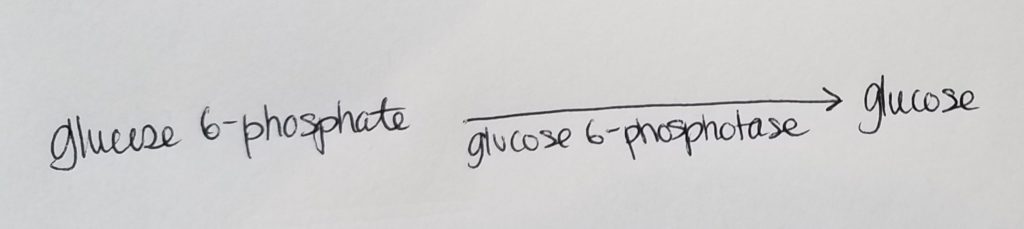
Cori Cycle (aka Lactic Acid Cycle).
Resources.
References.
Bean, J. (2019). ETC & oxidative phosphorylation.
Bean, J. (2019). Glycolysis and gluconeogenesis.
Bean, J. (2019). TCA cycle.
Ferrier, D. (2017). Biochemistry (7th ed.). Philidelphia, PA: Lippincott Illustrated Reviews.
Lieberman, M., & Peet, A. (2017). Marks’ basic medical biochemistry: A clinical approach(5th ed.). Philadelphia, PA: LWW.
Last edited: 08.08.2019
See Appendix A Amino acid abbreviations; Appendix B Glucogenic, Ketogenic, and Glucoketogenic Amino acids; Appendix C 20 Amino Acids to Know Drawn at Physiological pH 7.4.
Review of Macromolecules and Functional Groups.
Condensation (dehydration) synthesis.
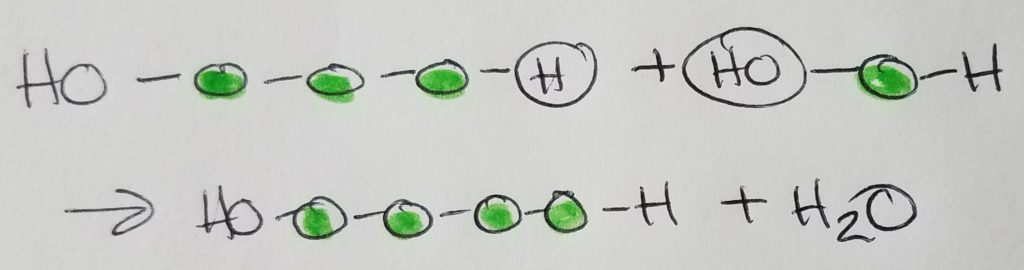
Hydrolysis.
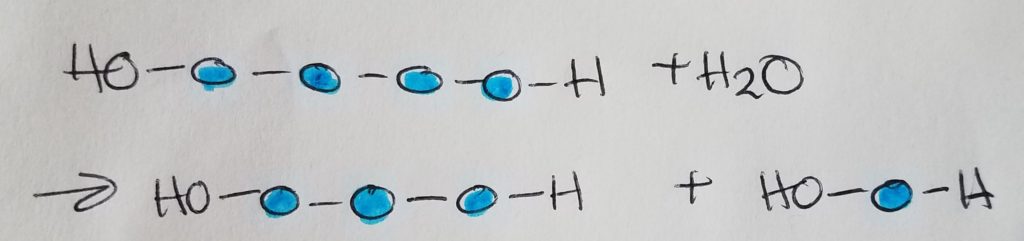
Carbohydrates.
A. Structure.
A sugar monomer is a single sugar unit called a monosaccharide. Two monosaccharides make up a disaccharide via glycosidic linkage. Sucrose = glucose + fructose. Maltose = glucose + glucose.
Glycosidic linkages are condensation/dehydration reactions in which H2O is a product (is produced in the reaction). The formula for a typical sugar is CnH2nOn.
Sugars may be aldoses (aldehyde group at top) or ketoses (ketone group at top). On a Fischer projection, L-sugars have the hydroxyl group on the left of the last chiral carbon (carbon farthest away from the aldehyde or ketone group). D-sugars have the hydroxyl group to the right of the last chiral carbon. The human body almost exclusively uses D-sugars.
Sugars can have the same molecular formula but still be different due to stereochemistry. Galactose and glucose can have the same molecular formula. They are different sugars because they are arranged differently. It’s like having 5 red legos and 5 blue legos; you can put them together in many different combinations.
B.Function.
Examples of polysaccharides: starch (plant energy storage), cellulose (fiber-like used for plant cell walls), glycogen (animals’s energy storage in muscles and liver), chitin (exoskeletons and fungal cell walls). Example function of carbohydrates and used for energy storage and structural support.
Proteins.
A. Structure.
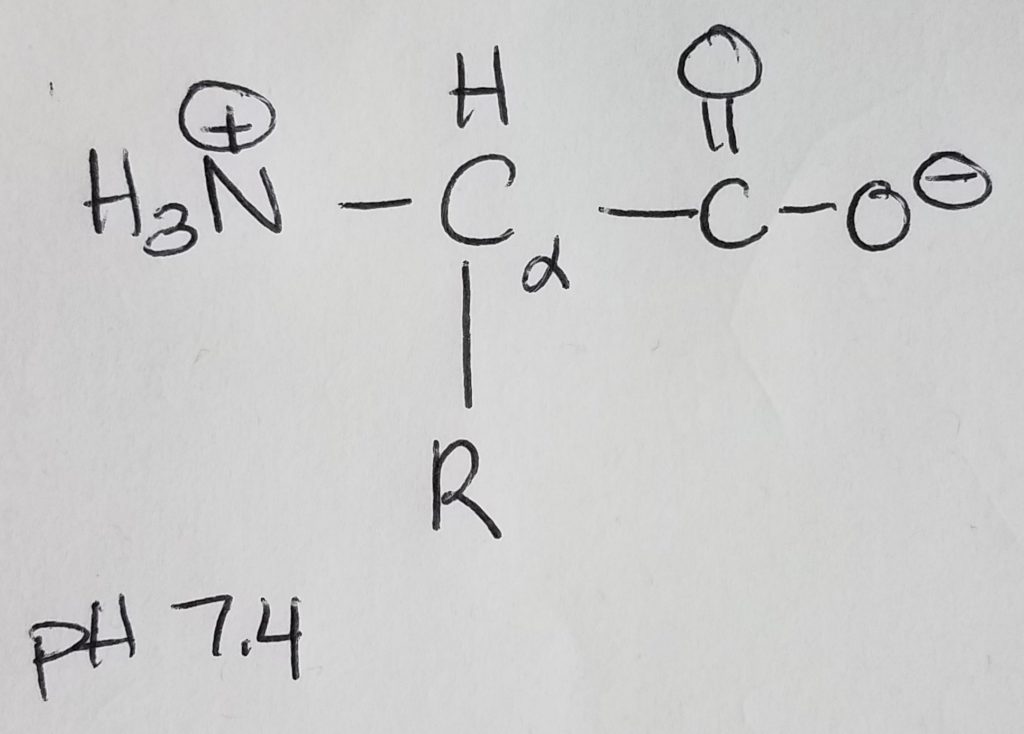
Amino acids are monomers for proteins which are 1+ polypeptide chains (polymer). 2 AA’s are joined via peptide bonding. Polypeptide chains can get longer and longer and you can put multiples of them together to form complex molecules. Once AA’s become part of a chain they are called “residues”. AA’s are made up of: an amine group, a carboxylic acid group, a hydrogen at the alpha-carbon, and a R-sidechain.
At physiological pH of 7.4, the amino group is protonated (+NH3) and the carboxylic acid group is deprotonated (COO-). Because the molecule has a dual charge, it’s called a zwitterion. The charged amino group at the leftmost position of the chain is called the N-terminus; the carboxylate ion at the rightmost position of the chain is called the C-terminus. AA’s are joined together to form a chain via peptide bonding.
Remember that if the pH < pKa, then that group will be protonated (e.g. +NH3, COOH). If pH > pKa, then that group will be deprotonated (e.g. NH2, and COO-). Physiological pH is 7.4; pKa is ~9.0 for amines; pKa is ~4.75 for carboxylic acid. Histidine’s pKa is very close but a little on the basic side at physiological pH.
Amino acids have stereochemistry (they have mirror images).
The “L” designation in L-amino acids denotes that the zwitterion is oriented with the protonated amine group at the left and the carboxylate ion on the right. The human body uses L-amino acids with the exception of glycine which is achiral. This is also important because the L stereochemistry is what our body uses in ligand-receptor binding (e.g. enzymes).
The alpha-carbon is the carbon directly bonded to the carboxylic acid group. The alpha amine is the amine group directly bonded to the alpha carbon.
Peptide bonding is when the carboxylate end of one AA covalently bonds with the charged amino end of another AA giving off a water molecule. Peptide bonding is a special form of dehydration synthesis (condensation reaction) between two amino acids. A more general term for peptide bonding is amide bonding. Bonding sequence matters. If amino A is bonded to amino B and then to amino C, the charged amino group of A is retained. In the order A-B-C, the carboxylate ion of amino C is retained. Bonding order matters. A-B-C is not the same as B-C-A.
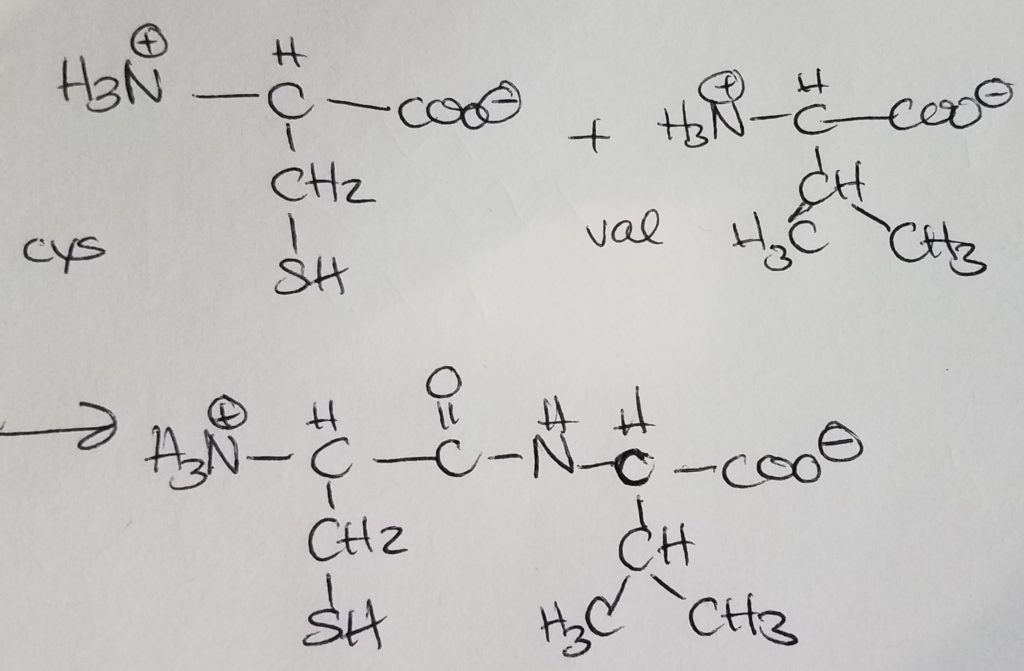
Proteins have primary, secondary, tertiary, and quaternary structures.
Primary structure is characterized by an unique sequence of amino acids in a polypeptide. Structure and function go hand in hand. Any tiny change in structure could impact the function of the protein.
Secondary structure is characterized by repeated folding of the polypeptide BACKBONE. The keyword is backbone. Hydrogen bonds (H-bonds) stabilize peptide bonds. The alpha helix and B-pleated sheets (parallel or antiparallel). Antiparallel configuration is more stable as steric and angle strain.
http://www.chem.ucla.edu/~harding/notes/strain_01.pdf
https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Alkanes/Properties_of_Alkanes/Cycloalkanes/Ring_Strain_and_the_Structure_of_Cycloalkanes
Tertiary structure is characterized by lots more globular folding and interactions between R-sidechains/groups (H bonds, ionic bonds, sulfhydral bonds, disulfide bridges, salt bridges). This is where the polar/nonpolar, acidic/basic, etc. properties of amino residues becomes an important consideration.
Quaternary structure is characterized by 1+ separate tertiary structures assembling together to form a more complex molecule. Often the “core” or pockets created may hold inorganic ions and some of the gaps created may be ligand-receptor sites. The act of binding and releasing can cause conformational changes altering the function of the protein.
B. Function.
Amino acids may be classified as essential (ones the human body cannot make and must come from exogenous sources). Nonessential amino acids are ones the human body can make.
Amino acids may be classified based on the polar/nonpolar characteristics of the sidechain: polar, nonpolar; acidic/basic/uncharged or neutral.
Amino acids may be classified based on aromatic or aliphatic (structural comparison).
Amino acids may be classified based on what they can make: glucogenic (make glucose); ketogenic (make ketones); glucoketogenic (make both glucose and ketones).
Proteins can have all sorts of functions: acting as enzymes, structural support, antibodies, cell signalling, transport, and movement.
See Appendix A Amino acid abbreviations; Appendix B Glucogenic, Ketogenic, and Glucoketogenic Amino acids; Appendix C 20 Amino Acids to Know Drawn at Physiological pH 7.4.
Lipids.
A. Structure.
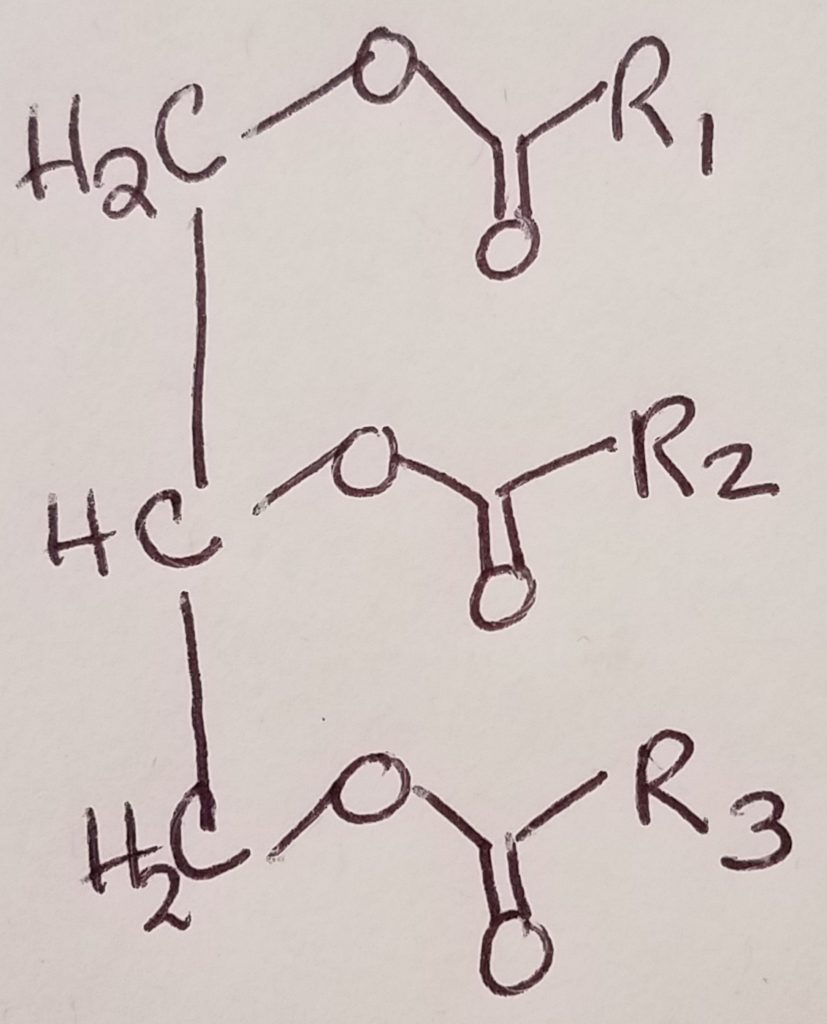
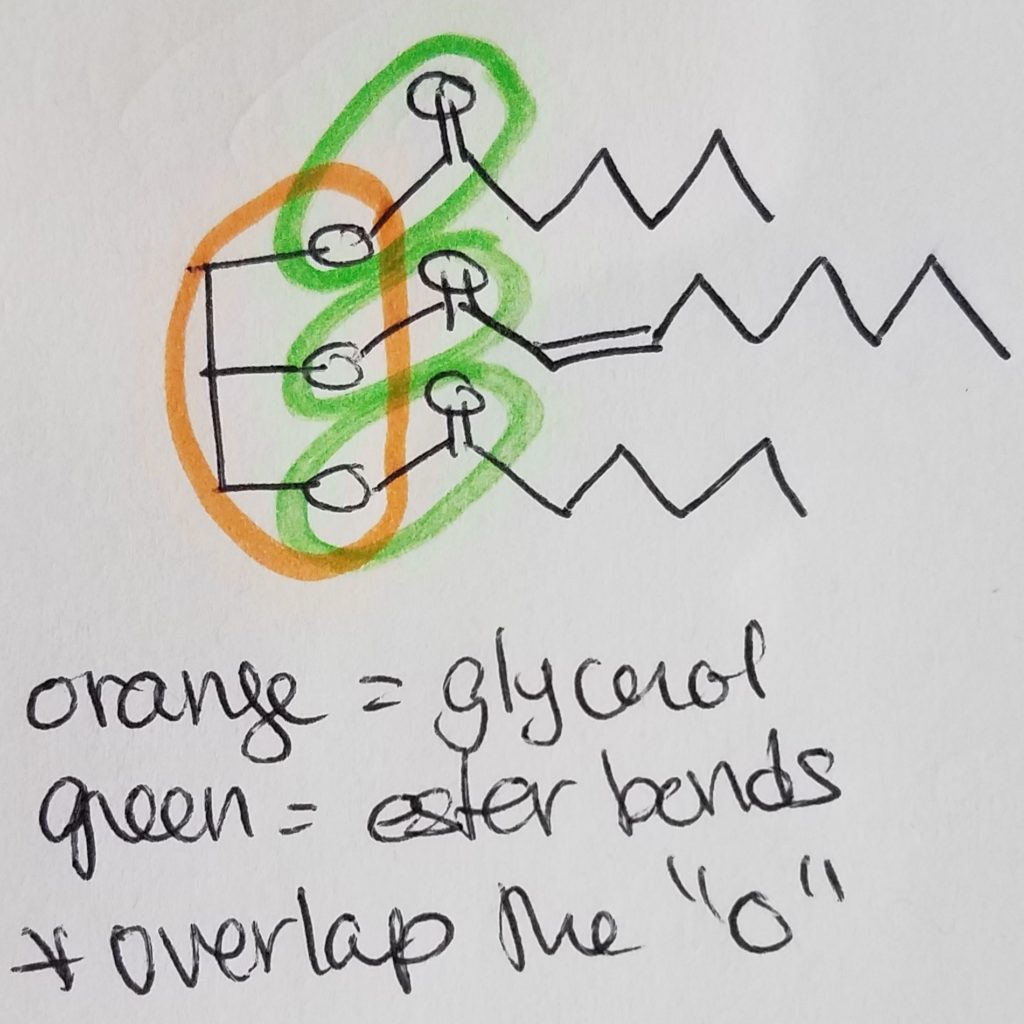
A functional unit of lipids is a triglyceride which is a glycerol (trihydric alcohol) and fatty acids (acyl groups which can be saturated or unsaturated long hydrocarbon with a carboxylic acid head. The carboxylate head bonds with the hydroxyl (of the glycerol) via ester linkages. The fatty acids that get attached to the glycerol do not have to be the same. Triglycerides are amphipathic (the key to this structure is its hydrophilic and hydrophobic duality).
Phospholipid = glycerol + 2 FA chains + phosphate + head-group. The key to this structure is its hydrophilic and hydrophobic duality. This is important to cell membranes, micelles, etc.
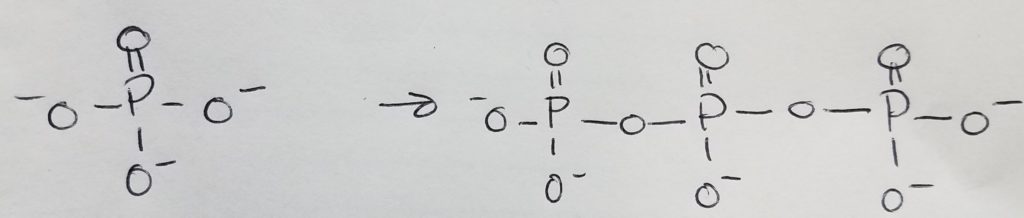
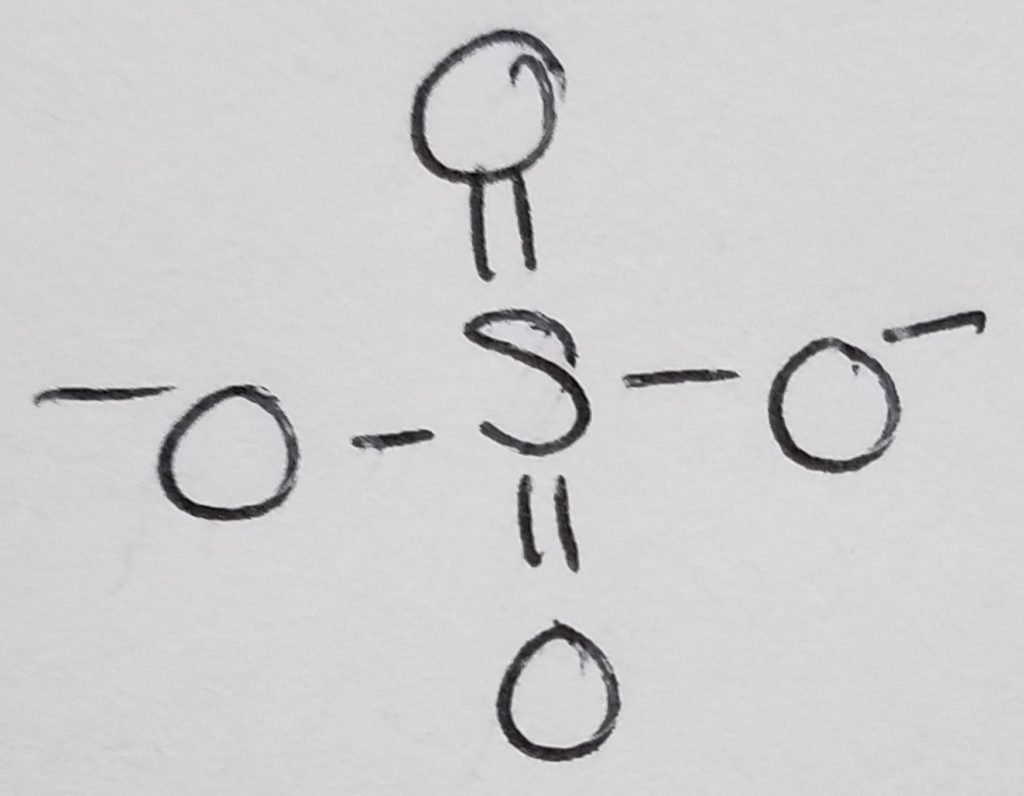
Waxes = 2 long hydrocarbon tails connected via ester bond.
Steroids = 4 carbon rings with no fatty acid tails.
http://www.biologie.ens.fr/~mthomas/L3/intro_biologie/2-sucres-lipides-acides-nucleiques.pdf
**Circle the ester bond(s) and circle the glycerol component. BE CAREFUL that the ester bonds will overlap with the glycerol oxygen.
B. Function.
Some functions include (but not limited to): creating a barrier; insulation; protection; energy storage; hormones, carriers (e.g. fat soluble vitamins) and signalling.
Nucleic Acids.
A. Structure
Nucleotides are the monomers for nucleic acids. A nucleotide = pentose sugar (e.g. deoxyribose or ribose) + phosphate (mono, di, or tri) + nitrogenous base. “Deoxyribose” just means that the second carbon in the ring does not have a hydroxyl group (i.e. carbon 2 is bonded to two hydrogens). A nucleoSide = pentose sugar (e.g. deoxyribose or ribose) + nitrogenous base = nucleoTide without the phosphates.
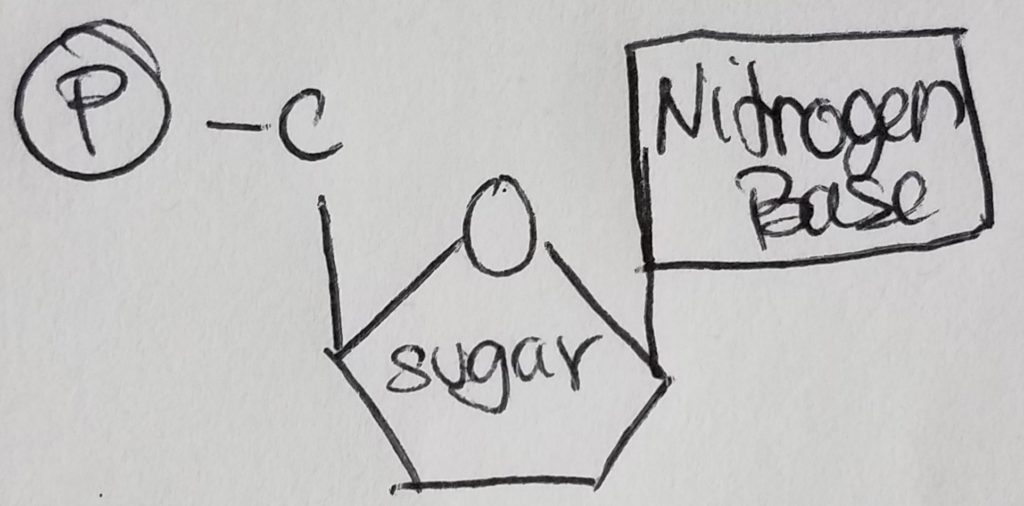
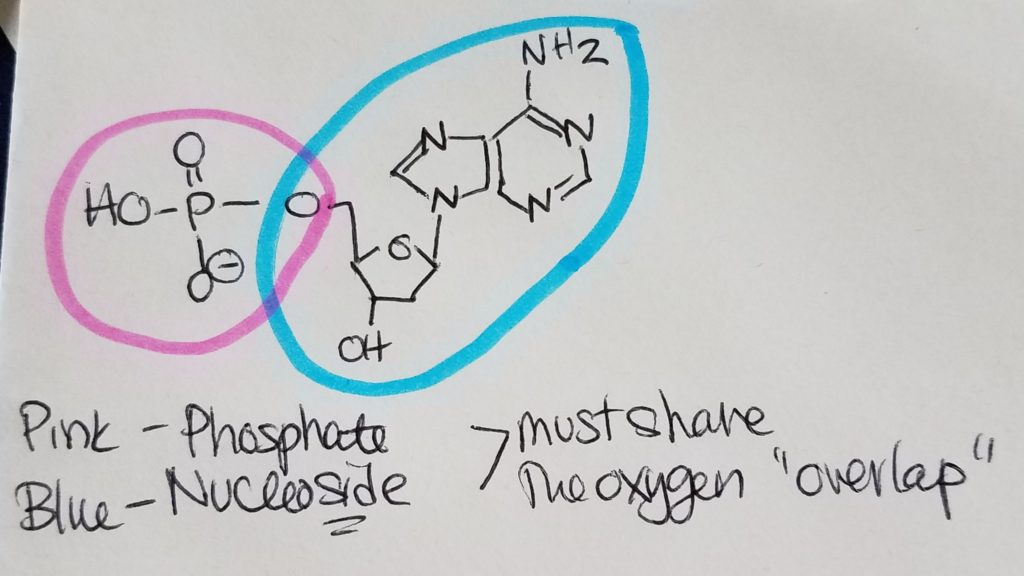
The pentose sugar is bonded to the nitrogenous base via N-glycosidic bond. The phosphates are bonded to the pentose sugar via phosphoester bonding.
https://chem.libretexts.org/Courses/Sacramento_City_College/SCC%3A_Chem_309_-_General%2C_Organic_and_Biochemistry_(Bennett)/Text/13%3A_Functional_Group_Reactions/13.10%3A_Phosphoester_Formation
The nitrogen bases used are guanine, adenosine, cytosine, thymine, and uracil (DNA pairs are C-G and A-T, RNA pairs are C-G and A-U). Adenine and guanine are purines (5 ring connected to a 6 ring); cytosine, thymine, and uracil are pyrimidines. Base pairs between C and G are stronger due to an extra H-bond.
How to tell them apart?
Cytosine has an amine group bonded to one of the carbons on the ring. Adenosine has a carbonyl group with the carbonyl carbon as part of the ring. Uracil has two carbonyl groups, each with the carbonyl carbon as part of the ring.
Adenine has an amine group hanging off the 6-ring structure; guanine has a carbonyl group with the carbonyl carbon as part of the ring structure.
The nucleotide monomers are connected via covalent bonding carbon 5 of one sugar to the carbon 3 of the other sugar (phosphate group sandwiched inbetween). We call this the 5′ (five prime) position and 3′ (three prime) position. It is a condensation reaction forming a phosphodiester bond between the sugars and phosphate groups (alternating sugar-phosphate-sugar-phosphate etc.) with water given off as a product.
Differences between DNA and RNA.
1. Sugar. DNA has deoxyribose (missing -OH on 2nd carbon); RNA has ribose.
2. Base pairs. DNA’s base pairs are C-G, A-T. RNA’s base pairs are C-G, A-U.
3. # of strands. DNA has two strands in a double helix configuration (H-bonds help to stabilize). RNA is single-stranded.
B. Function.
Nucleic acids are in genetic material like DNA and RNA.
The nitrogen bases used are guanine, adenosine, cytosine, thymine, and uracil (DNA pairs are C-G and A-T, RNA pairs are C-G and A-U). Adenine and guanine are purines (5 ring connected to a 6 ring); cytosine, thymine, and uracil are pyrimidines. Base pairs between C and G are stronger due to an extra H-bond.
How to tell them apart?
Cytosine has an amine group bonded to one of the carbons on the ring. Adenosine has a carbonyl group with the carbonyl carbon as part of the ring. Uracil has two carbonyl groups, each with the carbonyl carbon as part of the ring.
Adenine has an amine group hanging off the 6-ring structure; guanine has a carbonyl group with the carbonyl carbon as part of the ring structure.
The nucleotide monomers are connected via covalent bonding carbon 5 of one sugar to the carbon 3 of the other sugar (phosphate group sandwiched inbetween). We call this the 5′ (five prime) position and 3′ (three prime) position. It is a condensation reaction forming a phosphodiester bond between the sugars and phosphate groups (alternating sugar-phosphate-sugar-phosphate etc.) with water given off as a product.
Differences between DNA and RNA.
1. Sugar. DNA has deoxyribose (missing -OH on 2nd carbon); RNA has ribose.
2. Base pairs. DNA’s base pairs are C-G, A-T. RNA’s base pairs are C-G, A-U.
3. # of strands. DNA has two strands in a double helix configuration (H-bonds help to stabilize). RNA is single-stranded.
B. Function.
Nucleic acids are in genetic material like DNA and RNA.
Central Dogma.
As far as our understanding today, scientists believe that genes determine the sequence of mRNA which in turn determines the sequence of proteins.
https://www.khanacademy.org/science/biology/gene-expression-central-dogma/central-dogma-transcription/a/the-genetic-code-discovery-and-properties
Basic Steps: From Gene to the Primary Structure of Protein.
Step 1. Transcription.
In the nucleus, RNA polymerase unzips DNA and reads DNA in order to make and spit out pre-mRNA (transcribed from the DNA) using nucleoside triphosphates as building materials. Emphasis on triphosphates because the process requires lots of energy which is available in those phosphate bonds (e.g. each time phosphate gets cleaved).
Step 2. Editing the pre-mRNA.
In the nucleus, the pre-mRNA needs to be edited. A methylated structure called a “cap” is placed on pre-mRNA’s 5′ end. A poly-adenosine monophosphate tail is added at the 3′ end. Introns are spliced out leaving only the exons.
https://www.cell.com/trends/biochemical-sciences/fulltext/S0968-0004(19)30002-7
Step 3. Translation.
The “edited/fixed-up” mRNA is transported out of the nucleus and into the cytoplasm at the rough endoplasmic reticulum. The mRNA gets sandwiched between the large and small ribosomal subunits. The ribosome is the assembly site. The tRNA is the shuttle that pairs its anticodon to the codon on the mRNA and brings in the correct amino acid for assembly. As the ribosome moves along reading the mRNA, the protein strand that’s being formed (queue-fashioned) starts to stick out of the ribosome. This step is called “translation” because it’s like decoding a secret message on the mRNA to make the protein that you want.
https://www.khanacademy.org/science/biology/gene-expression-central-dogma/translation-polypeptides/v/translation-mrna-to-protein
See Appendix A Amino acid abbreviations; Appendix B Glucogenic, Ketogenic, and Glucoketogenic Amino acids; Appendix C 20 Amino Acids to Know Drawn at Physiological pH 7.4.
Appendix A. Amino acid abbreviations.
Appendix B. Glucogenic, Ketogenic, and Glucoketogenic Amino acids.
Ketogenic (LK): lysine, leucine.
Ketoglucogenic (ITFWY): isoleucine, threonine, phenylalanine, tryptophan, tyrosine.
Glucogenic: alanine, arginine, asparagine, aspartate, cysteine, glutamate, glutamine, glycine, histidine, methionine, proline, serine, valine.
Appendix C. 20 Amino Acids to Know Drawn at Physiological pH 7.4
Glycine
The only achiral amino acid because the sidechain is just a hydrogen.
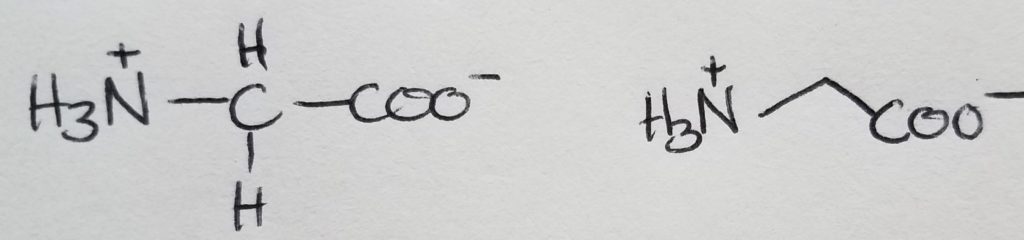
Alanine
The shortest sidechain, methyl group.
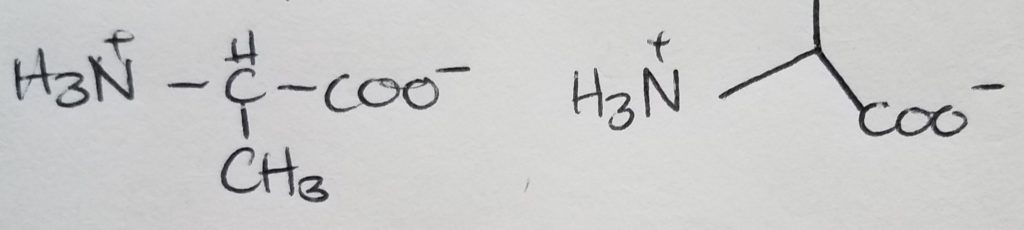
Valine
Looks like a short “V”.
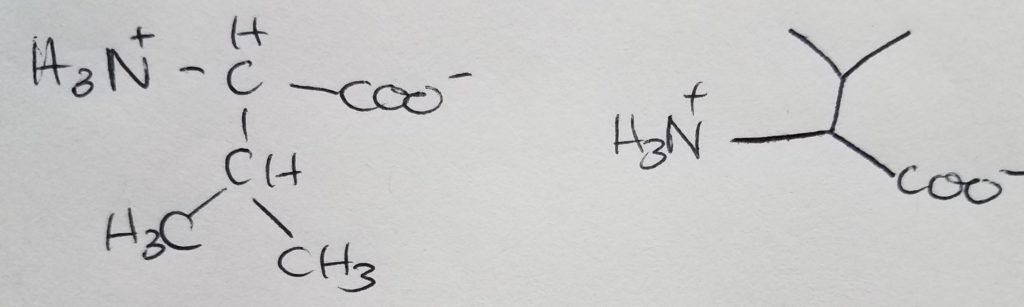
Isoleucine
Isomer of leucine.
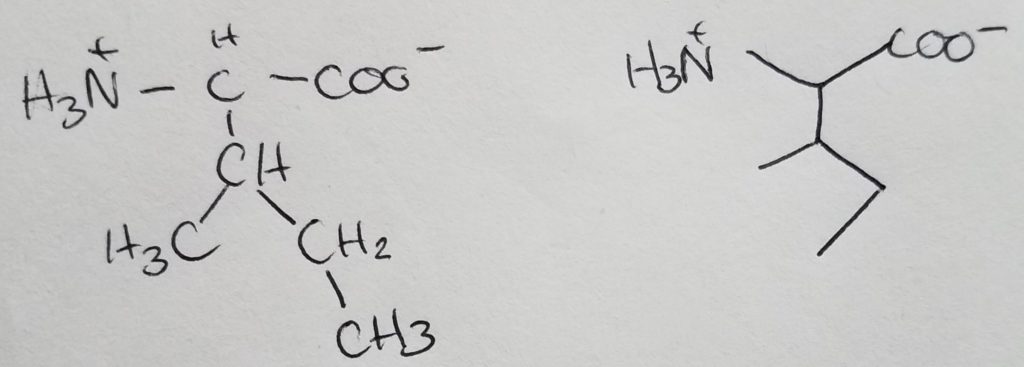
Leucine
Isomer of isoleucine.
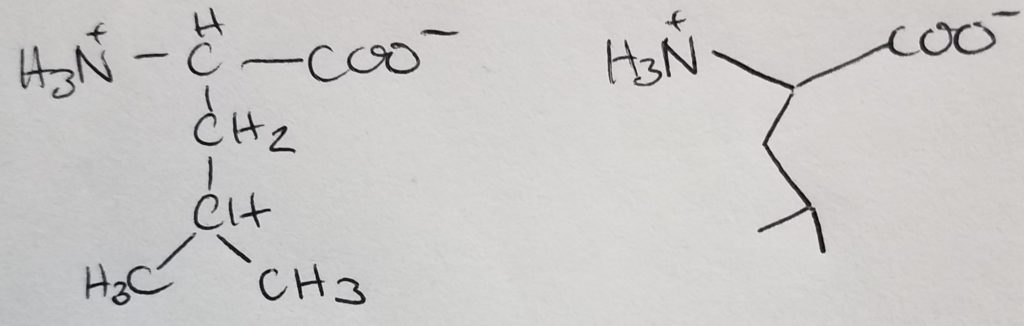
Resources.
References.
Bean, J. (2019). Amino.
Bean, J. (2019). Macromolecules.
Bean, J. (2019). Nucleotides and nucleic acids.
Bean, J. (2019). Protein structure.
Ferrier, D. (2017). Biochemistry (7th ed.). Philidelphia, PA: Lippincott Illustrated Reviews.
Bean, J. (2019). Nucleotides and nucleic acids.
Bean, J. (2019). Protein structure.
Ferrier, D. (2017). Biochemistry (7th ed.). Philidelphia, PA: Lippincott Illustrated Reviews.
Lieberman, M., & Peet, A. (2017). Marks’ basic medical biochemistry: A clinical approach(5th ed.). Philadelphia, PA: LWW.
[Study guide covering chapter 7, PDF]
Last edited: 08.05.2019
Characteristics of 3D Structure.
Requirements for 3D Structure.
3D Structure of Peptide Bond.
Secondary Structure.
Tertiary Structure.
Quaternary Structure.
Quantication of Ligand Binding.
Structure-Function in Myoglobin and Hemoglobin.
Structure-Function Relationships in Immunoglobin.
Protein Folding.
Resources.
References.
Lieberman, M., & Peet, A. (2017). Marks’ basic medical biochemistry: A clinical approach(5th ed.). Philadelphia, PA: LWW.
[Study guide covering chapter 6, PDF]
Last edited: 08.05.2019
Structure.
Classification of Amino Acids.
Variations in Protein Structure.
Modified Amino Acids.
Resources.
References.
Lieberman, M., & Peet, A. (2017). Marks’ basic medical biochemistry: A clinical approach(5th ed.). Philadelphia, PA: LWW.
[Study guide covering chapter 5, PDF]
Last edited: 08.04.2019
Important functional groups: those with oxygen, nitrogen, phosphorus, sulfur.
Carbohydrates (sugars).
Lipids.
Nitrogen Compounds.
Free Radicals.
Oxidation/Reduction.
Acid/Base.
Bond Polarity & Partial Charges.
Resources.
References.
Lieberman, M., & Peet, A. (2017). Marks’ basic medical biochemistry: A clinical approach(5th ed.). Philadelphia, PA: LWW.
